
- Academic Editor
The tumor microenvironment, especially the extracellular matrix (ECM), plays a critical role in cancer initiation and progression, although its underlying mechanisms remain incompletely understood. Conventional therapies (such as chemotherapy, surgery, and radiotherapy) often produce unsatisfactory outcomes. Immunotherapy, while showing limited clinical success to date, holds considerable promise. Growing evidence indicates that the biophysical properties of the ECM interact with immune cells, contributing to mechanisms of immunotherapy resistance in cancer. Alterations in these ECM properties can impair immune cell infiltration and function, thereby diminishing the effectiveness of immunotherapeutic approaches. This review explores how the biophysical features of the ECM and their crosstalk with tumor immune evasion pathways highlight the potential of ECM-targeted immunotherapy as an innovative strategy for cancer treatment.
The tumor microenvironment (TME) represents a pivotal determinant in tumor progression and the development of resistance to therapeutic interventions, particularly in its capacity to facilitate immune evasion and confer resistance to immunotherapy. TME encompasses a diverse array of components, including peripheral immune cells, blood vessels, extracellular matrix (ECM), fibroblasts, lymphocytes, inflammatory cells originating from bone marrow, and signaling molecules [1, 2]. Among these components, ECM not only functions as a structural scaffold for tissues but also delivers essential biochemical and biomechanical signals that influence cell behavior, such as proliferation, survival, migration, and differentiation. Furthermore, the ECM regulates vascular development and immune responses [3, 4]. ECM is an intricate, three-dimensional macromolecular network primarily composed of collagen, non-collagenous glycoproteins, elastin, proteoglycans, and glycosaminoglycans (GAGs) [5, 6]. Structurally, ECM can be divided into the intermediate matrix and the basement membrane, each exhibiting distinct functions, compositions, and locations [7, 8]. The intermediate matrix forms a permeable 3D scaffold that surrounds cells, linking them to each other and to the basement membrane. In contrast, the basement membrane serves as a specialized ECM component that separates the epithelium from the surrounding matrix [9, 10]. Through continuous interactions with tumor cells, immune cells, and cancer-associated fibroblasts (CAFs), ECM influences tumor progression, metastasis, immune modulation, and metabolic reprogramming, highlighting its crucial role in cancer biology [11, 12, 13, 14] (Fig. 1).
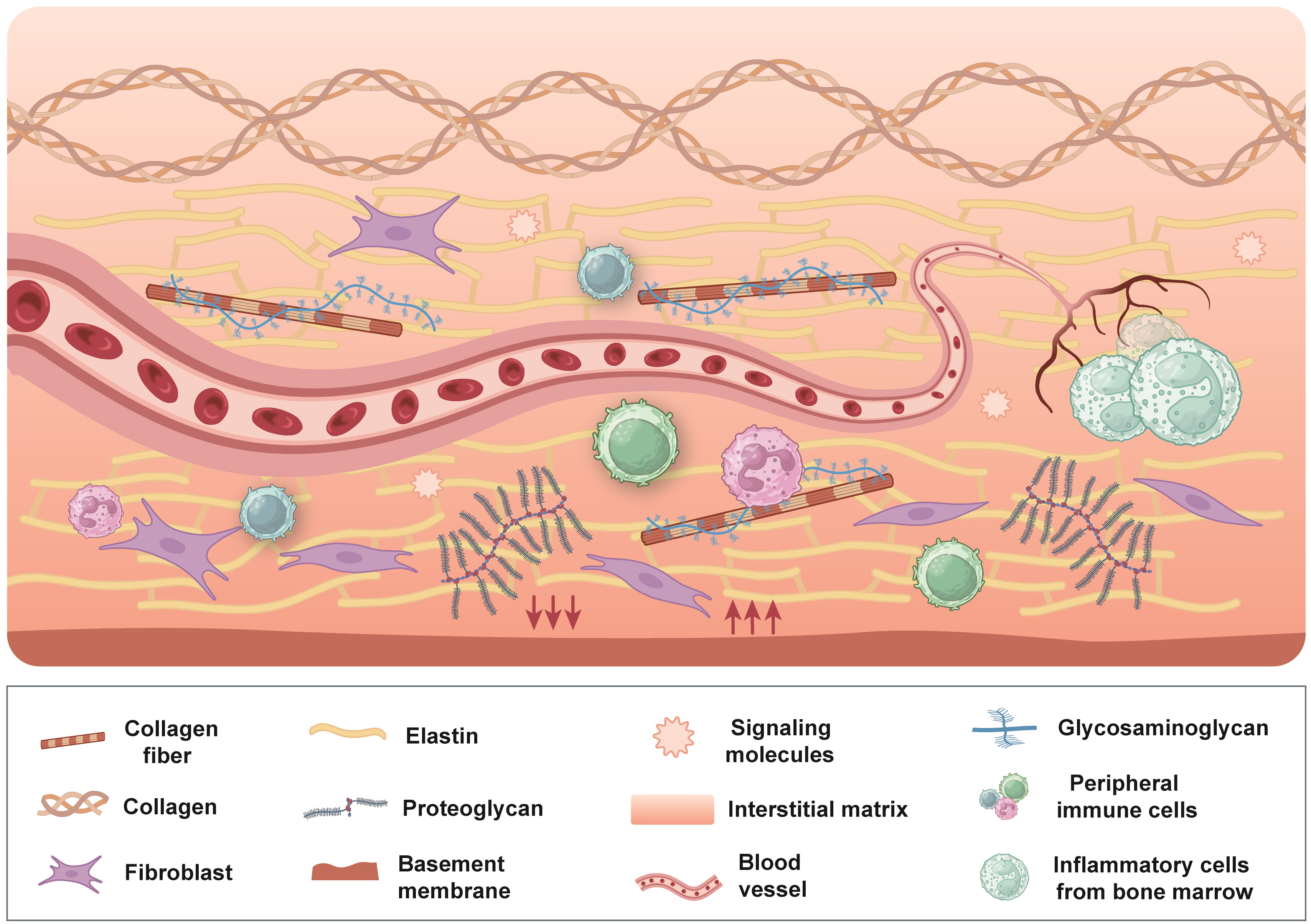 Fig. 1.
Fig. 1.
Overview of the tumor microenvironment. The tumor microenvironment is a multifaceted and dynamic environment comprising peripheral immune cells, blood vessels, fibroblasts, lymphocytes, bone marrow-derived inflammatory cells, signaling molecules, and ECM. ECM forms a highly intricate three-dimensional macromolecular framework primarily composed of collagen, non-collagenous glycoproteins, elastin, proteoglycans, and glycosaminoglycans, and is traditionally categorized into the intermediate matrix and the basement membrane. Through continuous and reciprocal interactions with tumor cells, immune cells, and cancer-associated fibroblasts, the ECM plays a pivotal role in shaping tumor progression, metastasis, immune evasion, and metabolic reprogramming. ECM, extracellular matrix. Figure created using Adobe Illustrator CC 2019.
The biophysical characteristics of ECM including stiffness, viscoelasticity, morphology, and permeability, are increasingly recognized for their significant influence on tumor behavior, with ECM stiffness emerging as a particularly critical factor. Post-translational modifications, such as hydroxylation and enzymatic cross-linking, can induce biomechanical alterations in the substrate, thereby activating mechanosensing pathways [15, 16]. Integrins, which are heterodimeric transmembrane receptors, play key roles in bidirectional signaling by linking to the intracellular actin cytoskeleton through interactions with ECM components. This process involves integrins detecting mechanical forces within the ECM and transmitting this information to intracellular proteins, such as focal adhesion kinase (FAK) and Src tyrosine kinase, a phenomenon known as mechanotransduction [17, 18, 19]. The interaction between ECM and cells through mechanotransduction is mediated by integrins binding to structural proteins and filamentous actin. The investigation into the dynamic relationship between ECM biophysical cues and intracellular signaling pathways has fueled the rapid advancement of the interdisciplinary field of cancer mechanobiology in recent years [20].
Furthermore, the biomechanical properties of ECM exert a significant influence on the immune cell behavior, presenting a promising direction for investigating the mechanistic regulation of the immune system [21]. The density and alignment of collagen and elastin are crucial determinants of ECM stiffness. Alterations in stromatogenesis and tumor mechanics are predominantly driven by the activities of CAFs, which modulate the composition, structure, and degradation of ECM, thereby contributing to increased stiffness [22, 23]. This increased stiffness not only impedes the immune cell infiltration but also disrupts the precise delivery of immunotherapeutic agents, while simultaneously activating signaling pathways that foster immunosuppression within the TME [24].
ECM is pivotal in modulating the tumor immune microenvironment (TIME) by recruiting and activating immune cells such as tumor-associated macrophages (TAMs), thereby fostering immunosuppression [25]. Additionally, ECM triggers metabolic reprogramming, resulting in the accumulation of lactate and the upregulation of indoleamine 2,3-dioxygenase (IDO), which collectively induce immune tolerance, suppression of cytotoxic T lymphocytes activity, and facilitation of tumor immune evasion [26, 27].
ECM remodeling and degradation are crucial in cancer progression. The well-known link between cancer and inflammation shows how chronic inflammation creates an environment that supports cancer cell growth, thus aiding tumor development [28]. Additionally, inflammation often causes tissue damage, which triggers tissue repair processes [29]. During this process, matrix metalloproteinases (MMPs) are essential in ECM degradation and restructuring, leading to the release of growth factors and signaling molecules that influence cell structure and behavior [30, 31]. Moreover, lysyl oxidase (LOX) promotes collagen deposition and cross-linking, forming a physical barrier that blocks T-cell infiltration into the tumor core [32]. This review explores how the biophysical characteristics of the ECM (such as stiffness, composition, and remodeling) affect tumor behavior, as well as how the ECM contributes to immune evasion and ECM-targeted immunotherapy. By gathering these insights, the review aims to guide the development of more effective treatments and advance cancer immunotherapy.
ECM is present in all organisms, exhibiting tissue-specific characteristics and undergoing continuous remodeling. This dynamic process involves various cellular activities such as synthesis, degradation, recombination, and chemical modification, which collectively maintain tissue homeostasis. The precise regulation of ECM remodeling mechanisms in vivo is crucial for normal physiological function. Disruptions in tumor cell behavior can lead to ECM imbalance at biochemical, biological, and structural levels. Alterations in ECM homeostasis, including modifications in ECM components and molecular organization, can alter its structure and biophysical properties, contributing to various pathological conditions. The biophysical properties of ECM, such as elasticity, matrix pore size, structure, and morphology, are essential for maintaining tissue homeostasis [33]. In the following subsections, we will explore ECM structure and biophysical characteristics from three perspectives: ECM components and structure, ECM stiffness and mechanotransduction, and ECM remodeling.
ECM serves as the primary structural framework maintaining balance within tissues and organs, predominantly made up of collagen, non-collagenous proteins, elastin, proteoglycans, and GAGs [34]. ECM components play critical roles in tumorigenesis and progression in the TME, where they influence immune responses. Tumor cells utilize surface receptors to interact with ECM components, triggering the activation of proteolytic enzymes that facilitate matrix degradation, thereby promoting tumor cell metastasis. The metastatic process in tumor cells is typically a result of the coordination of multiple pathways, rather than a single pathway, which includes ECM remodeling, degradation of collagen and elastin, and increased expression of tenascin and hyaluronic acid (HA).
Collagen constitutes a major component of ECM, accounting for approximately 90% of the human body’s ECM and 30% of its total protein content [35]. Collagen is an insoluble fibrous protein with a diameter of 1.5 nm, characterized by a triple helix structure formed by three parallel polypeptide alpha chains. Collagen is classified into 28 types based on variations in gene coding and peptide chain combinations. Type I, III, V, and VI collagen primarily exist in the interstitial matrix, while type IV collagen is predominantly found in the basement membrane [36, 37]. Collagen provides structural integrity and mechanical support to the ECM. LOX catalyzes collagen crosslinking to form a stable collagen network. However, overexpression of LOX can lead to excessive collagen cross-linking, creating a physical barrier that impedes immune cell infiltration [38]. The linear structure formed by collagen fibers in the tumor supports tumor cell migration [39]. Furthermore, collagen fibers play a key role in providing structural support to the ECM and play an important role in signal transduction [40, 41].
Non-collagenous glycoproteins play distinct roles and exhibit specific distributions in the ECM, with major components including fibronectin (FN), laminin (LN), and tenascin (TN) [42]. FN is a “V”-shaped dimer composed of two subunits connected by disulfide bonds, featuring multiple binding sites for collagen, heparin, and cellular components [43, 44]. Abnormal FN levels in the TME may influence tumor cell growth, differentiation, and migration [45]. LN, predominantly found in the basement membrane, is crucial for its development and stability [46]. TN, which is widely distributed in the TME, is often upregulated under conditions of tissue injury, inflammation, and cancer. Elevated TN levels are associated with poor prognosis in various cancers, like breast cancer and colorectal cancer [47]. In addition to their structural roles in the ECM, non-collagenous glycoproteins are involved in vital biological processes such as cell growth, differentiation, and signal communication [48]. Therefore, understanding the structure and function of non-collagen glycoproteins is of great significance for advancing cancer treatment.
Elastin is a water-insoluble protein composed of four primary amino acids: glycine, proline, alanine, and valine, typically arranged in a recurring pattern of 3 to 9 amino acids [49, 50]. Elastin is widely distributed in tissues such as blood vessels, skin, and cartilage, where it helps maintain tissue elasticity. It achieves its remarkable elasticity through the cross-linking of protoelastin (elastin precursor), forming a three-dimensional elastic network. This structure allows elastin to rapidly return to its original shape after being stretched and released multiple times [51]. ECM remodeling leads to altered tissue elasticity, inducing tumor migration and invasion [52]. Elastin-like polypeptides (ELPs) are derivatives of pro-elastin, synthesized based on the amino acid sequence of elastin. ELPs contain repetitive sequences such as Val-Pro-Gly-Xaa-Gly (VPGXG) [53]. During ECM remodeling, ELPs are often secreted into the extracellular microenvironment, potentially contributing to cancer progression and metastasis [54]. Preventing elastin degradation may block positive inflammatory feedback in tumors. Additionally, ELP as a tumor therapeutic carrier may offer new avenues for treatment.
Proteoglycans are important components of the ECM and consist of GAGs and core proteins. GAGs are covalently bonded to core proteins and are chain polymers consisting of repeating disaccharides. Due to their negative charge, GAGs can attract and trap cations, thereby absorbing water in the ECM [55, 56]. There are six types of GAGs in humans: chondroitin sulfate (CS), dermatan sulfate (DS), heparin sulfate (HS), keratan sulfate (KS), heparin, and HA [57]. Except for HA, all GAGs contain sulfate groups. Abnormal expression and synthesis of proteoglycan play a significant role in cancer progression. For instance, HA is frequently overexpressed in tumors and contributes to tumor development and survival by binding to receptors such as CD44 on the surface of cancer cells [58, 59]. HA can also interact with TAMs and regulatory T cells (Tregs), which impede the immune response to tumors [60, 61, 62].
The elastic force gradually exerted by ECM on cells is known as matrix stiffness, also referred to as Young’s modulus—defines ECM resistance to deformation. Research has indicated that ECM rigidity increases with age [63]. The stiffness of ECM is affected by its composition, the degree of collagen crosslinking, and proteoglycan modification [64]. It varies greatly across tissues, from less than 100 Pa to several MPa [65]. Tumor cells can stimulate stromal cells to release paracrine factors that promote cancer growth and metastasis. One crucial step in the transformation of the precancerous microenvironment is the conversion of normal stromal fibroblasts into CAFs known as fibroblast corruption [66, 67]. Both tumor cells and CAFs express ECM-modifying enzymes, such as LOX, which promote collagen crosslinking, thus increasing ECM stiffness and facilitating tumor cell proliferation [68]. Collagen undergoes extensive deposition and posttranslational crosslinking, forming a linear and reticular structure [69]. As collagen deposits and crosslinks over time, the ECM stiffens, driving cancer progression [70]. ECM components and stiffness can also transmit mechanical signals that activate cell membrane receptors and mechanical sensors (such as integrins, Piezo1, and TRPV4), initiating mechanical transduction [71] (Fig. 2).
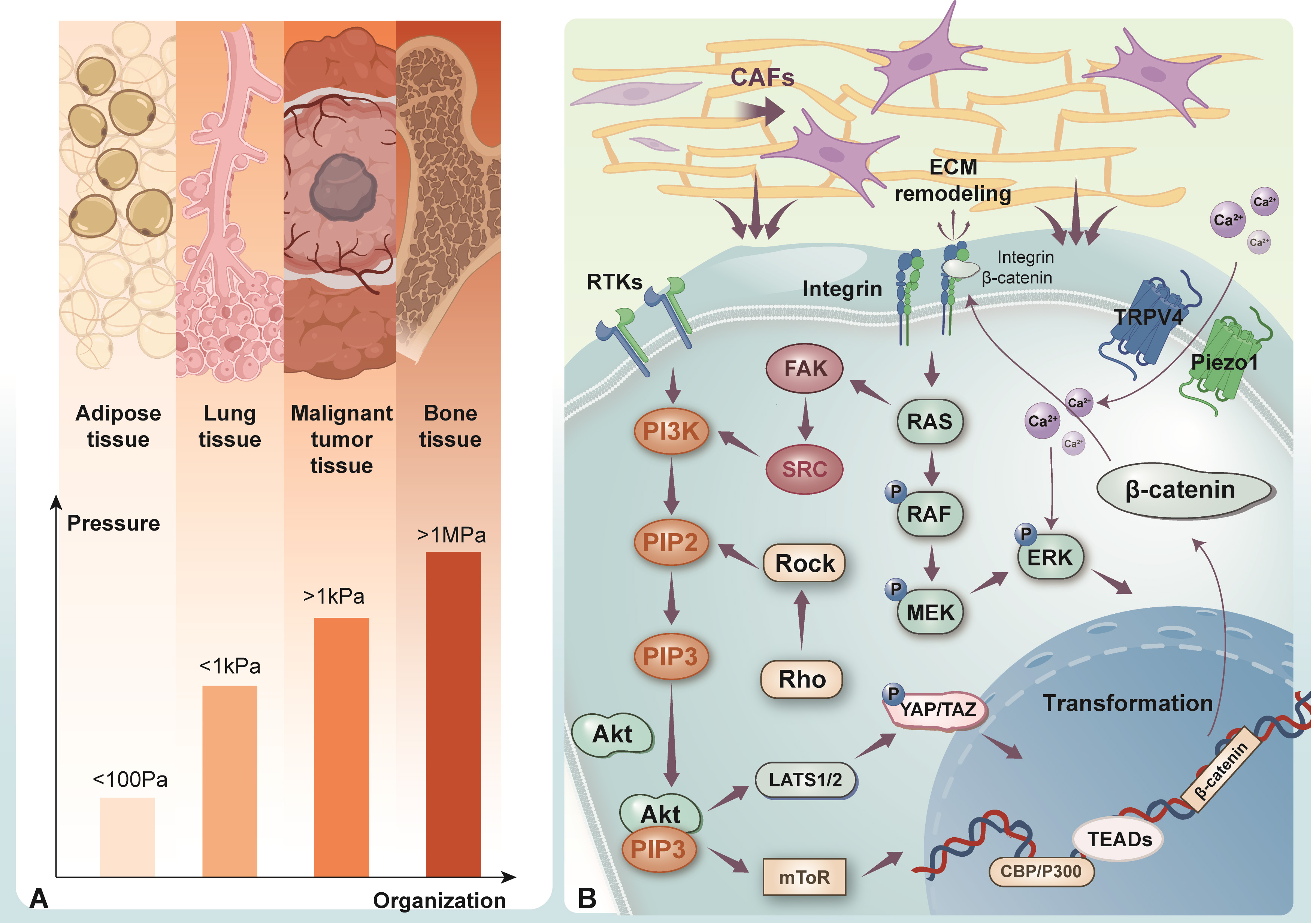 Fig. 2.
Fig. 2.
ECM stiffness in different tissues and mechanotransduction. (A) The stiffness of ECM exhibits variation across different tissues, spanning a range from less than 100 Pa to more than 1MPa. (B) ECM components convey mechanical signals through mechanosensitive receptors, including integrins, Piezo1, and TRPV4, thereby initiating mechanotransduction pathways. Integrins transmit mechanical forces and activate downstream effectors such as FAK, Src, and the Rho/ROCK pathway. Piezo 1 detects ECM stiffness and mediates calcium influx, which enhances cell migration, matrix remodeling, and cancer cell invasion. TRPV4 plays a key role in EMT by promoting nuclear translocation of Akt and YAP/TAZ, both critical for cellular transformation. ECM, extracellular matrix; FAK, focal adhesion kinase; Src, SRC proto-oncogene, non-receptor tyrosine kinase; Rho, Rhodopsin; ROCK, Rho associated coiled-coil containing protein kinase 1; TRPV4, transient receptor potential cation channel subfamily V member 4; EMT, epithelial-mesenchymal transition; Akt, protein kinase B; YAP, Yes-associated transcriptional regulator; TAZ, transcriptional coactivator with PDZ-binding motif; CAFs, cancer-associated fibroblasts; ERK, extracellular signal-regulated kinase; Ras, rat sarcoma virus; PI3K, phosphoinositide 3-kinase; RTKs, receptor tyrosine kinases; RAF, rapidly accelerated fibrosarcoma; MEK, mitogen-activated extracellular signal-regulated kinase; PIP2, phosphatidylinositol 4,5-bisphosphate; PIP3, phosphatidylinositol 3,4,5-trisphosphate; LATS1/2, large tumor suppressor kinase 1 and 2; TEADs, TEA domain transcription factors. Figure created using Adobe Illustrator CC 2019.
Integrins are a diverse group of transmembrane dimers composed of alpha and beta subunits, playing a crucial role in cell movement, growth, and cancer invasion [72, 73]. Integrins mediate cell-ECM interactions, converting ECM mechanical stimuli into intracellular signaling cascades. As key mediators between cells and ECM, integrins transmit mechanical forces through aggregation and conformational changes, thus activating downstream signaling molecules such as FAK, Src, and Rho/ROCK [74, 75]. Piezo1 is a mechanosensitive ion channel that detects ECM stiffness, triggering downstream signaling and increasing calcium ion concentrations [76, 77]. Piezo1 can also facilitate cell movement and enhance matrix restructuring, which, in turn, promotes cancer cell migration and invasion [78]. Another important mechanosensitive ion channel is TRPV4, which promotes epithelial-mesenchymal transition (EMT) by promoting Akt activation and the nuclear translocation of YAP/TAZ [79]. YAP/TAZ are key transcriptional regulators and nuclear effectors in the Hippo pathway, and their abnormal activation contributes to tumor proliferation and alterations in the TME [80]. When ECM stiffness is low, YAP/TAZ exist in the cytoplasm. However, mechanical cues such as increased ECM rigidity, tension, and cellular concentration induce the relocation of YAP/TAZ from the cytoplasm to the nucleus [81, 82]. In the nucleus, YAP/TAZ interact with TEA domain family proteins (TEADs), modulating the expression of specific genes facilitating cancer progression and metastasis [83, 84]. YAP/TAZ detect physical forces within the cells and participate in mechanotransduction, regulating essential cellular activities such as growth, specialization, and stem cell maintenance [85, 86]. ECM is a highly dynamic, non-cellular structure that maintains close contact with cells, providing structural support while maintaining tissue balance through continuous remodeling. Remodeling, degradation, or processing of ECM can induce dynamic changes in cell and tissue behavior, influencing fundamental biological functions such as cell movement, shape, growth, and survival [87, 88]. In cancer, uncontrolled cell growth leads to ECM reshaping. ECM breakdown typically occurs via two primary mechanisms: intracellular and extracellular (Fig. 3). Intracellular breakdown involves lysosomal proteases within phagolysosomes degrading ECM, while extracellular remodeling is mediated by proteases that are released into the environment [89, 90]. TAMs and CAFs play critical roles in the extracellular remodeling of tumorigenic ECM. TAMs release proteases that facilitate the breakdown and formation of collagen fibers surrounding the tumor boundary [91, 92, 93]. The elevation of ECM stiffness can trigger downstream signaling pathways via mechanotransduction, thereby inducing the secretion of MMPs by cancer cells and stromal cells. The enhanced activity of MMPs not only accelerates ECM degradation and remodeling but also compromises the basement membrane structure, which serves as a critical barrier to cancer cell migration and invasion. This disruption provides physical channels that facilitate the invasive migration of tumor cells, ultimately enhancing their proliferative capacity and metastatic potential [71, 94]. Moreover, as the primary producers of MMPs, cancer-associated fibroblasts (CAFs) break down ECM constituents and reorganize their protein architecture, thereby facilitating directional migration and invasion of cancer cells within the stiffened ECM microenvironment [95]. In summary, MMPs play a pivotal role in tumor progression and metastasis by regulating ECM remodeling, promoting angiogenesis, and enhancing cell migration and invasion [96]. Changes in the ECM are also driven by mutant fibroblasts releasing growth factors, ultimately directly altering ECM components [97]. ECM remodeling promotes tumor progression by modifying mechanical properties such as elasticity and stiffness and by enhancing the availability of growth factors and cytokines [98, 99]. Additionally, ECM remodeling contributes to treatment resistance and tumor progression by increasing intratumoral interstitial fluid pressure (IFP), which impedes drug perfusion and limits intratumoral distribution [17, 100, 101].
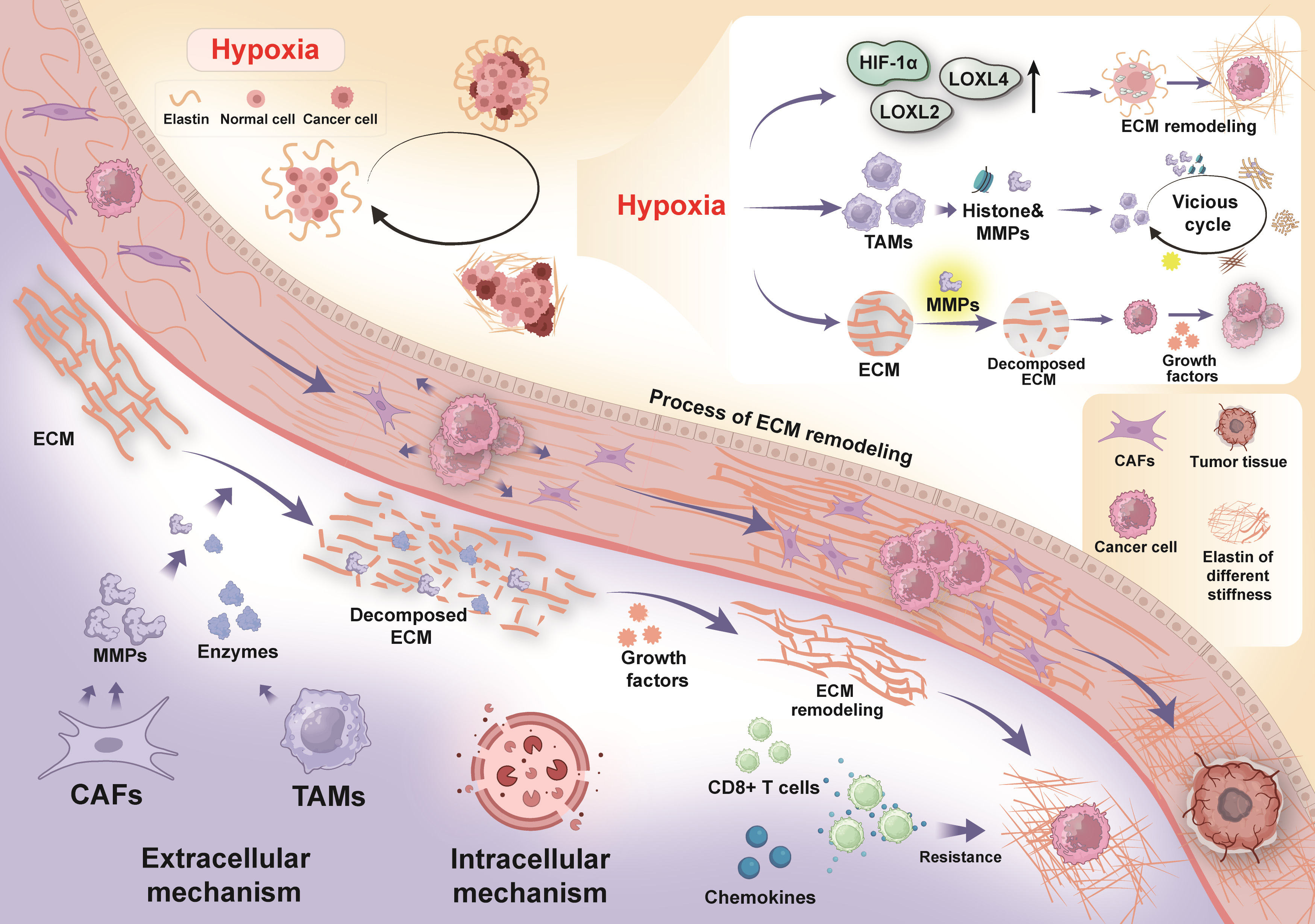 Fig. 3.
Fig. 3.
ECM remodeling. The degradation of ECM occurs through both
intracellular and extracellular mechanisms. Intracellularly, ECM breakdown is
facilitated by lysosomal proteases within phagolysosomes, while extracellular
remodeling is mediated by proteases that are released into the surrounding
environment. CAFs secrete MMPs that drive the degradation of the ECM. TAMs also
release proteases that promote the breakdown and reconstruction of collagen
fibers. ECM remodeling not only alters the physical structure of the tissue but
also enhances the availability of growth factors and cytokines. The degradation
of ECM subsequently triggers the secretion of chemokines, which, in turn, elevate
the tumor cell resistance to CD8+ T cells. Furthermore, hypoxia induces collagen
crosslinking and further ECM remodeling by upregulating the expression of LOX and
its homologues, such as LOXL2 and LOXL4, as well as HIF-1
Hypoxia induces ECM remodeling, promoting inflammation and increasing the
heterogeneity of TME [102]. The stiffness of ECM can lead to the vasculature
compression, worsening hypoxia, raising intra-vascular pressure, and interfering
with the diffusion of nutrients, oxygen, and metabolic products [103, 104].
Hypoxia enhances the expression of hypoxia-inducible factors (HIFs), which, in
turn, increase the transcription of downstream factors [105]. Hypoxia induces
collagen crosslinking and ECM remodeling through upregulating the expression of
LOX and its homologues (e.g., LOXL2 and LOXL4), as well as HIF-1
ECM remodeling plays a critical role in regulating tissue homeostasis and cellular behavior. In cancer, ECM remodeling often works in conjunction with increased ECM stiffness to alter the state of the TME, thereby driving tumor malignancy. Inhibiting the activity and production of ECM remodeling enzymes may offer strategies for immunotherapy.
ECM plays a pivotal role in tumor progression and immune evasion by facilitating immunosuppression, modulating biochemical signaling pathways, interacting with immune cells, and altering the tumor metabolic microenvironment. Understanding how ECM contributes to immune escape through these mechanisms could provide novel insights for advancing tumor immunotherapy (Fig. 4).
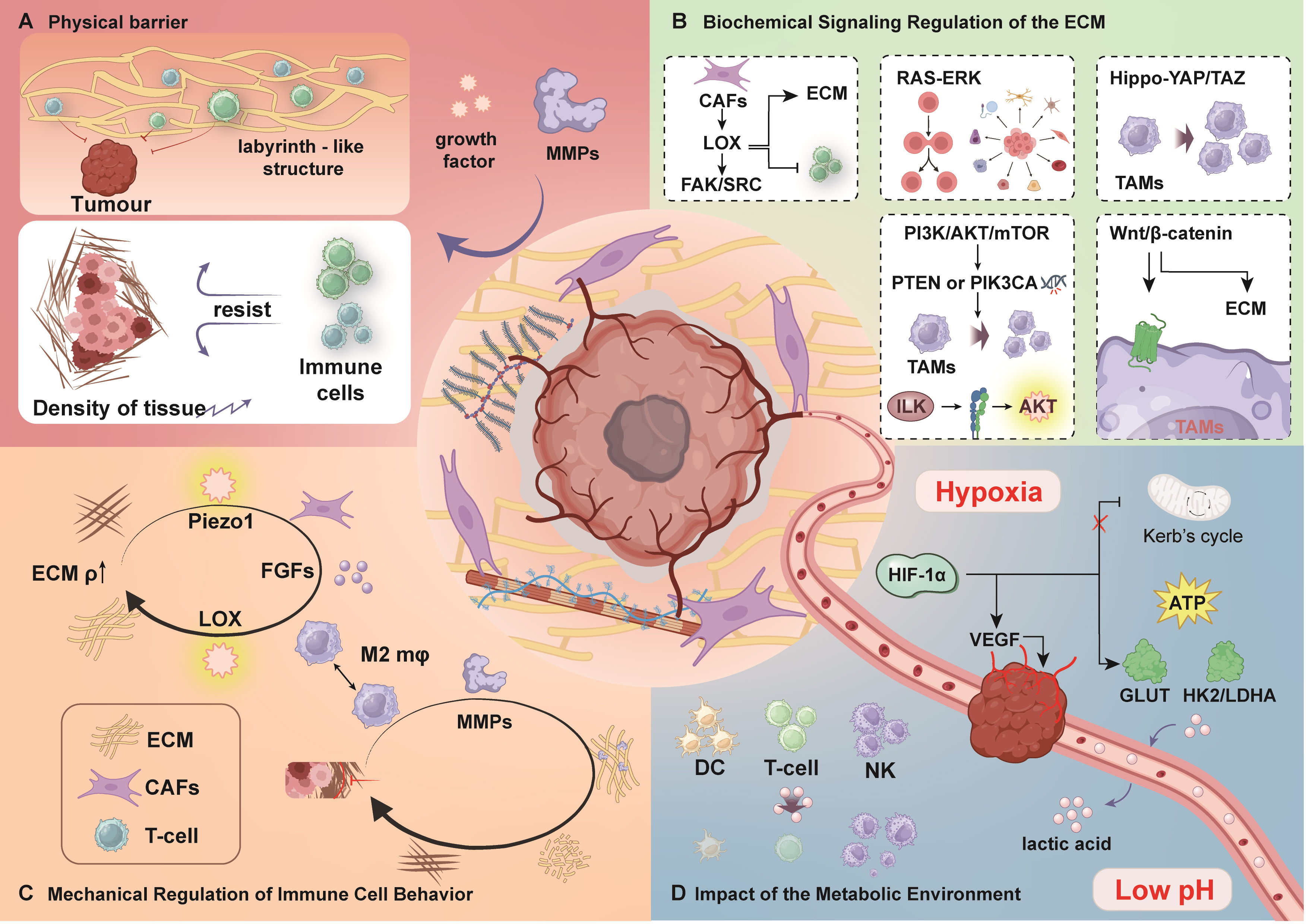 Fig. 4.
Fig. 4.
ECM and immune evasion. (A) Physical barrier. Tumor cells and
CAFs frequently upregulate LOX, which enhances ECM rigidity through cross-linking
of collagen and elastin fibers. Meanwhile, MMPs disrupt ECM structure, resulting
in increased stiffness and density that collectively impede immune cell
infiltration. (B) Biochemical signaling in the ECM. The FAK/Src pathway
contributes to cell survival and growth, whereas the RAS-ERK pathway regulates
proliferation and differentiation. The Hippo pathway promotes the recruitment of
TAMs, thereby facilitating immune evasion. The PI3K/Akt pathway supports cell
survival, and the Wnt/
ECM plays a crucial role in the formation of an immunosuppressive
microenvironment, which is essential for tumor progression and immune evasion.
The cytokines and chemokines released by the immunosuppressive system can result
in immune evasion [111]. Abnormal remodeling of the ECM creates an
immunosuppressive microenvironment that impedes the anti-tumor activity of the
immune system in targeting and killing cancer cells [112]. Additionally, ECM
serves as a storage reservoir for growth factors, cytokines, and chemokines,
which contribute to the regulation of immune cell functions associated with
tumors [113]. In the TME, stromal cells differentiate into CAFs, and both tumor
cells and CAFs often overexpress LOX, which catalyzes the covalent cross-linking
of elastin and collagen fibers, resulting in increased ECM stiffness [70, 114].
Increased ECM stiffness raises the CD4+ cells/CD8+ T cells ratio and impairs
their activation [107, 115]. Moreover, macrophages in the TME are transformed
into immunosuppressive M2-type macrophages, which is promoted by CAFs activation
through the secretion of transforming growth factor-
CAFs contribute significantly to the immune suppression in the TME. One of their primary mechanisms involves the inhibition of T-cell infiltration, which occurs through the C-X-C ligand 12 (CXCL12)-regulated T cell rejection and ECM-mediated T cell trapping. This process limits the number of immune cells capable of effectively engaging with the tumor. Additionally, an elevated density of collagen and the activation of Piezo1 promote macrophage polarization, ultimately resulting in a reduction in T cell numbers [123]. In the TME, the accumulation of Tregs, TAMs, and myeloid-derived suppressor cells (MDSCs) contributes to the establishment of a tumor-specific immunological microenvironment [124]. The accumulation of Tregs and M2-type macrophages accentuates immunosuppressive effects, thereby diminishing the ability of immune cells to eradicate tumors [125]. CAFs are implicated in several pro-tumorigenic activities, including the production of ECM components, angiogenesis, and the promotion of tumor growth [126]. Additionally, CAFs are contributed to the establishment of an immunosuppressive microenvironment that deteriorates immune surveillance, potentially facilitating cancer proliferation and progression [127]. Programmed cell death protein 1 (PD-1) is expressed in most activated immune cells, with its ligand programmed cell death ligand 1 (PD-L1) counteracting immune surveillance and further promoting immune evasion by the tumor [128]. The increased ECM stiffness associated with hypoxia in the TME adversely affects the functional capacity of cytotoxic T lymphocytes, as well as Tregs, further contributing to the suppression of anti-tumor immunity.
Dysregulated remodeling of the ECM contributes significantly to the establishment of a physical and biochemical barrier that impairs the infiltration of immune cells into tumors, thereby promoting immune escape. Targeting the ECM to normalize its structure and function is emerging as a promising strategy for improving tumor treatment. By restoring the balance in the ECM, it may be possible to enhance immune cell infiltration and improve the effectiveness of immunotherapy. One avenue of research involves the use of antifibrotic drugs, which are being explored for their potential to reverse ECM remodeling. These drugs aim to reduce ECM stiffness and modify its components, making it more conducive to immune cell activity. This approach could potentially improve the response to immunotherapies, such as checkpoint inhibitors, by enabling better immune cell penetration and function in the TME.
ECM serves as both a physical and biochemical barrier that impedes immune cell
penetration and regulates the function of various immune cells in the TME [129].
Furthermore, the features of ECM structure and mechanics play a role in a complex
biochemical signaling process that creates an immunosuppressive microenvironment
in the tumor [130]. Biochemical signaling pathways are triggered by the
interaction with ECM molecules and ECM-bound growth factors. The structure and
mechanical features of ECM, as external signals, activate a cascade of signal
transducers. Once the signal is sensed by cancer cells, several important
elements on the cell membrane, known as mechanosensing elements, are triggered,
including FAK, integrins, syndecan, and calmodulin, to initiate an unusual force
transmission from the ECM to the nucleus with uneven force transmission [107, 131, 132]. Additionally, multiple cytokines (e.g., caveolin-1 and TGF-
The interplay between the Hippo pathway and other key signaling cascades, such
as Wnt, Notch, PI3K/Akt, and TGF-
During tumorigenesis, a bidirectional regulatory relationship exists between ECM and immune cells. The composition of the ECM is controlled by immune cells to maintain and regulate the healthy stromal environment [164]. The physical and molecular characteristics of ECM govern the growth, activation, and mobility of immune cells within tissues, thus generating an immunosuppressive tumor immune microenvironment (TIME) [165]. The immune cell populations in the TIME can be broadly classified into two categories: antitumoral immune cells and intratumoral immune suppressive cells. Antitumoral immune cells include T cells, macrophages, lymphocytes, natural killer (NK) cells, and dendritic cells (DCs), whereas myeloid-derived suppressor cells (MDSC), Tregs, and M2-type macrophages are classified as immunosuppressive cells [166, 167].
Studies have shown that ECM regulates T cell recruitment and aggregation while inhibiting T-cell proliferation and differentiation, thus facilitating immune escape of tumors [113, 168]. In addition to influencing tumor cell proliferation, migration, and invasion, the ECM influences immune cell adhesion and migration, as well as modulating inflammation in the TME [169]. Versican, one of the components of ECM, stimulates and activates pre-tumorigenic immune cells (e.g., macrophages), thereby promoting the induction of tumor inflammation [170]. The abnormality of ECM stimulates the increase in intracellular YAP expression levels and the levels of fibroblast, inducing fibroblast activation and differentiation into CAFs, a hallmark of poor cancer prognosis [20, 171, 172]. CAFs secrete fibroblast growth factors that stimulate macrophage polarization towards the M2-type, and they also release WNT-2, which inhibits dendritic cell differentiation and activation [173, 174]. This immunosuppressive environment is reinforced by the presence of Tregs and M2-type macrophages, thus inducing a positive feedback loop [175]. Tumor-associated macrophages constitute half of the immune cells in the TME and can be activated under various stressful conditions [176].
Most TAMs can polarize into M1 and M2 phenotypes. Increased ECM stiffness hinders dendritic cell maturation and promotes the transition of macrophage to the M2 phenotype [71, 177]. Piezo1, a mechanosensitive protein, activates M2-type macrophage polarization [78]. M2-type macrophages can secrete MMPs (e.g., MMP7, MMP9, and MMP12) and LOX, creating a physical barrier that excludes T cells, thereby reducing the effectiveness of immunotherapy, especially for immune checkpoint inhibitors (ICIs), whose efficacy relies on the presence of T cells in the tumor core [88, 178, 179].
ECM fibers can regulate cell migration trajectories and limit the interaction between immune cells and cancer cells. Additionally, ECM contributes to the establishment of the immunosuppressive microenvironment by attracting Tregs and promoting M2-type macrophage polarization [175, 180, 181]. Inhibition of LOX has been shown to enhance T-cell infiltration and increase sensitivity to immune checkpoint inhibitors [182]. ECM releases cytokines and chemokines that facilitate the extravasation and aggregation of T cells in the tumor [183]. Tumor cells downregulate the expression of major histocompatibility complex (MHC) molecules, inhibiting antigen presentation and facilitating immune evasion [184, 185]. HIFs in ECM can impair the cytotoxic effector function of NK and CD8+ T cells by upregulating the ectonucleotidases, CD39 and CD73 [186, 187]. HA can support immune cell function by interacting with CD44, influencing the migration and localization of leukocytes, thereby affecting immune surveillance against tumors [188]. The role of ECM components in modulating tumor-immune interaction and regulating immune cell functions in the TME has gained increasing recognition. The interactions between ECM and immune cells shape the TME, which in turn regulate the recruitment and function of immune cells, ultimately influencing tumor development and clinical outcome [189]. Immune cell behavior is governed by the mechanical properties of ECM, where physical characteristics such as stiffness, collagen density, and alignment of collagen fibers directly impact immune cell infiltration, function, and their interaction with tumor cells [190]. The regulation of immune cell function by ECM stiffness is a dynamic process, where immune signals can also alter ECM stiffness through multiple mechanisms as part of the adaptive response. An elevated concentration of collagen is an important factor influencing immune cell distribution and function in the TME by altering their movement and positioning. For example, ECM stiffness stress increases the proportion of CD4+/CD8+ T cells [107]. However, this mechanical stress can concurrently impair the function of CD8+ T cells, leading to alterations in their surface markers, subpopulation distributions, and gene expression, which in turn affects their tumor-fighting ability [191]. Furthermore, the structural arrangement of collagen fibers can create a physical barrier that hinders the infiltration of CD8+ T cells into the tumor [192, 193]. The degradation of ECM can trigger the release of chemokines, which further enhance the tumor cell resistance to CD8+ T cells, undermining their ability to eliminate tumor cells [20, 194, 195]. While the modulation of ECM-immune interactions has shown promise, the development of immunotherapies targeting the ECM remains an area requiring further attention. For instance, the inhibition of LOX or MMPs can render ECM, thereby enhancing the efficacy of ICIs, as demonstrated in pancreatic cancer models. However, certain patient populations, such as those with hepatocellular carcinoma accompanied by cirrhosis, may experience detrimental effects, including tissue damage (e.g., vascular leakage), due to the excessive degradation of ECM components. Furthermore, ECM “dependence” is highly context-dependent, with varying tumor responses to ECM-targeted therapies: breast cancer, characterized by elevated collagen content, may exhibit a distinct therapeutic response compared to glioblastoma, which possesses low collagen levels and is enriched in HA [6, 196]. Consequently, the development of personalized therapeutic strategies that incorporate ECM-specific biomarkers, such as circulating collagen fragments, HA levels, and YAP translocation, in conjunction with advanced “local delivery” techniques, such as nanoparticle-based targeting of tumor ECM, is crucial for optimizing treatment outcomes.
The metabolic environment in the TME significantly affects tumor cell proliferation and promotes immune escape. In 1930, Otto Warburg discovered the Warburg effect in cancer cells, observing that tumors exhibit increased glucose uptake and excess lactate formation even under normoxic conditions [197, 198]. Although acidic hypoxic conditions are typically nonviable for most cells, tumors are highly adaptable and can still grow normally in such an environment [199]. In low-oxygen environments, aggressive tumors rapidly generate ATP through glycolysis to meet their energy requirements [200]. Cancer cells tend to preferentially utilize aerobic glycolysis rather than mitochondrial oxidative phosphorylation (OXPHOS), despite OXPHOS being more efficient in terms of ATP production from a single glucose molecule [201]. Additionally, elevated lactate levels resulting from glycolysis lower the extracellular pH, which not only promotes ECM degradation but also impairs antigen presentation by DCs and suppresses T-cell activity [202, 203].
Hypoxia plays a pivotal role in tumor metabolism. As tumor growth accelerates
with increasing tumor solid stress, compressing tumor and lymphatic vessels leads
to a hypoxic state [204, 205]. The combination of hypoxia and the heterogeneous
TME with elevated inflammation remodels the immune landscape by activating
multiple signaling cascades, ultimately resulting in immunosuppression [206, 207]. Hypoxia recruits immune cells, facilitating the migration of
tumor-infiltrating immune cells to the peripheral regions of the tumor, thereby
promoting tumor invasion [208]. Hypoxia fosters cellular proliferation and
survival by activating the PI3K/Akt and MAPK signaling pathways [209, 210].
Additionally, hypoxia enhances the secretion of TGF-
Chemotherapy, radiation therapy, and surgery represent conventional modalities for cancer treatment, each characterized by inherent limitations and associated side effects. In contrast, immunotherapy offers a promising alternative, often yielding more favorable therapeutic outcomes. However, immunotherapy does not exhibit efficacy across all types of tumors, with certain “cold” tumors exhibiting suboptimal responses. Cancer cells can escape from immune surveillance through various mechanisms, including the activation of pathways such as the PD-1/PD-L1, CTLA-4/B7, and IDO [218, 219]. Future approaches may involve stimulating immune activity in tumors devoid of immune infiltration through different antigen-presenting strategies [220]. Immunotherapy is broadly classified into three principal categories: active immunotherapy, passive immunotherapy, and combination immunotherapy [221]. Active immunotherapy seeks to invigorate the patients’ immune system, enabling it to specifically target and eradicate tumor cells through the activation of the autoimmune response [222]. Conversely, passive immunotherapy involves the use of tumor cytokines, monoclonal antibodies, and immune cells to kill tumor cells [223]. The forefront of clinical practice is currently marked by combination immunotherapy, which integrates immunotherapy with both targeted therapies and chemotherapy [224]. Furthermore, immunotherapy can be delineated into non-specific immunotherapy (such as cytokine therapy and NK cell therapy) and tumor-specific immunotherapy (such as DC-based immunotherapy and T-cell therapy) [225]. Despite notable achievements in tumor immunotherapy, challenges such as tumor immune tolerance and immune insensitivity remain unresolved. This section delineates present immunotherapy strategies targeting ECM, emphasizing ECM stiffness, ECM remodeling enzymes, immune checkpoint blockers, chimeric antigen receptor (CAR)-T cells, and the formidable associated challenges with these approaches. These strategies aim to enhance the effectiveness of immunotherapy with a focus on ECM (Table 1, Ref. [2, 89, 226, 227, 228, 229, 230, 231, 232, 233, 234, 235, 236, 237, 238, 239, 240, 241, 242, 243, 244, 245, 246, 247, 248, 249, 250, 251, 252, 253, 254, 255, 256, 257, 258, 259, 260]).
| Therapy strategies | Therapeutic targets | Mechanisms | Effects | References |
| Targeting ECM stiffness | Collagen, HA, signaling pathways (PI3K/Akt/mTOR, RASERK, Wnt/ |
Direct destruction of collagen using collagenase; reduction of collagen production by inhibitors such as Hsp47; reduction of ECM stiffness using LOX inhibitors such as |
Reduce ECM stiffness, improve drug penetration, promote anti-tumor immune cell infiltration, and inhibit tumor cell proliferation. | [226, 227, 228, 229, 230, 231, 232, 233, 234, 235, 236, 237] |
| ECM remodeling enzymes | MMPs, ADAMTS, LOX, FAP, Cathepsin B (CTSB). | Development of drugs that inhibit the activity of MMPs (e.g., minocycline); Baicalein, NDGA, and Zileuton can combat tumors by inhibiting LOX activity; CAR T-cell therapy targeting FAP; inhibition of the production and function of related enzymes. | Inhibit excessive ECM degradation and abnormal remodeling, reduce tumor cell migration and invasion, enhance immune cell infiltration, and improve immunotherapeutic sensitivity. | [2, 89, 238, 239, 240, 241, 242, 243, 244, 245] |
| Immune checkpoint blockade in the ECM | Immune checkpoint, such as PD-1/PD-L1, CTLA-4, etc. | Utilization of CTLA4- and PD1-targeting drugs to block the ligand-receptor axis and activate T-cell immune responses. | Enhance the body’s immune response to tumor cells and inhibit the growth and spread of tumor cells. | [89, 245, 246, 247, 248, 249, 250, 251, 252, 253, 254] |
| CAR-T cell and ECM | Tumor antigens, immune checkpoint inhibitors. | Collection of patient T cells genetically modified to express CAR and infused back into the body to recognize and kill tumor cells; combined with immune checkpoint blockade therapy. | Improve the recognition and killing ability of CAR-T cells on tumor cells, break through the tumor immune escape, especially for early-stage solid tumors, which is expected to improve the treatment response and survival rate. | [255, 256, 257, 258, 259, 260] |
Tumor growth, metastasis, and immune evasion are strongly linked to the rigidity
of ECM [261]. Targeting ECM components such as collagen and HA can facilitate the
infiltration of anti-tumor immune cells into the tumor microenvironment [226].
Collagen, as the primary component contributing to ECM rigidity, plays a critical
role in this context. The direct degradation of collagen through the application
of collagenases reduces ECM stiffness, thereby enhancing drug penetration into
the tumor [227, 228]. Strategies aimed at collagen degradation have shown
potential in preclinical studies. For instance, collagen-associated inhibitors,
like Hsp47 (a collagen-specific chaperone protein), have been used as inhibitors
to reduce collagen production and decrease ECM rigidity [229, 230, 231]. Preclinical
studies have shown that
ECM stiffness can transmit mechanical forces through ECM mechanoreceptors (e.g.,
FAK, integrins, syndecan, and calreticulin), which convey mechanical signals to
the cell interior [264]. Tumor proliferation can be effectively inhibited by
blocking key signaling pathways implicated in tumor growth, such as
PI3K/Akt/mTOR, RAS-ERK, and Wnt/
Enzymes that degrade ECM include MMPs and a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS) [238]. MMPs play roles in ECM remodeling and can enhance tumor invasion, angiogenesis, and metastasis [239]. MMPs modify ECM dynamics and communication pathways by breaking down ECM, leading to the liberation of growth factors and cytokines trapped within ECM, consequently changing its mechanical properties and signaling functions [240]. For example, secreted factors can bind to membrane receptors in stromal and epithelial cells, activating Src, which enhances malignant cycling by potentiating epithelial-stromal interactions [272, 273]. Moreover, MMPs promote cancer cell proliferation through neoangiogenesis [108]. LOX is associated with tumor progression and metastasis, and compounds such as Baicalein, NDGA, and Zileuton have been shown to inhibit LOX activity, combating tumor growth [2, 241]. Tissue inhibitory factors of MMPs (TIMPs) suppress MMP activity, and the imbalance between MMPs and TIMPs is closely associated with tumor progression [274]. Conventional therapies tend to kill tumor cells directly, but most patients die of tumor metastasis [275, 276]. As important ECM-degrading enzymes, MMPs represent potential targets for drug development. Inhibiting the activity of MMPs can help to inhibit tumor progression and improve the efficacy of tumor immunotherapy [275]. For example, the drug Minocycline can inhibit MMP activity by suppressing MMP-9 mRNA transcription and protein activity [242].
ADAMTS degrades ECM proteoglycans and promotes macrophage invasion, ultimately
triggering inflammation [277]. Fibroblast activation protein-
ECM-degrading enzymes are essential to remodel the ECM and influence its biophysical properties. Targeting the production and function of these enzymes represents an effective strategy for cancer treatment. This approach could reduce ECM stiffness, enhance the immune cell infiltration, and increase the sensitivity of cancer cells to immunotherapy. The combination of immunotherapy with a targeted inhibition of ECM-degrading enzyme holds promising potential for improving cancer immunotherapy outcomes.
The discovery of immune checkpoints and their inhibitors marked a significant milestone in medical oncology. They regulate the immune system and prevent breakdown in self-tolerance, thereby protecting peripheral tissues [246]. By targeting specific target proteins, the proliferation, growth, and spread of cancer cells can be impaired [247]. Tumors can upregulate immune checkpoint proteins, enabling tumor cells to evade immune surveillance [248]. Immune checkpoint inhibitors (ICIs) treatment blocks the ligand-receptor axis and triggers anti-tumor effect by activating the T-cell immune response [249]. ICIs targeting PD-1 and CTLA-4 are the most discussed [250]. The PD-1 protein and its partner PD-L1 receptor form an important immune checkpoint pathway with high clinical efficacy in patients with solid tumors [251]. In the absence of the tumor ligand of PD-L1 (which binds to PD-1), the T-cell response becomes less inhibited towards tumor cells [252]. By blocking immune cells, immune checkpoint blockade (ICB) therapy hinders the inhibition of T cells and renders patients’ immune systems more active against tumors [253]. Drugs targeting CTLA-4 and PD-1 are currently in clinical use as ICIs [254]. Notably, the efficacy and resistance mechanisms of ICIs vary across cancer types due to tumor-specific microenvironmental features: Non-small cell lung cancer (NSCLC): PD-L1 expression level is a key biomarker; tumors with high PD-L1 expression generally respond better to PD-1/PD-L1 inhibitors. However, not all NSCLC patients show a positive benefit and positive antitumor response to immunotherapy due to various factors, like the expression rate of PD-1/PD-L1. Targeted therapy has emerged as the first-line treatment for NSCLC patients harboring gene mutations that drive sensitivity, including prevalent oncogenic mutations such as EGFR, ALK, and ROS1 [278, 279]. For example, a high tumor mutation burden (TMB) generates abundant neoantigens, increasing T-cell recognition and sensitivity to ICIs in melanoma [280]. Moreover, high levels of MDSCs and M2-type macrophages secrete IL-10 and arginase-1, which deplete T cells and reduce their effector functions in bladder cancer [27, 281]. Reduction of CAFs has been shown to correlate with the improved ICIs effectiveness in PDAC, while the immunosuppressive function of CAFs contributes to the resistance of PDAC in a mouse model and facilitates tumor immune evasion [282, 283]. For example, a high tumor mutation burden (TMB) produces many neoantigens, increasing T-cell recognition and responsiveness to ICIs in melanoma [280]. Furthermore, high levels of MDSCs and M2 macrophages secrete IL-10 and arginase-1, which deplete T cells and diminish their effector functions in bladder cancer [27, 281]. Reducing CAFs has shown a correlation with improved efficacy of ICIs in PDAC, while the immunosuppressive function of CAFs contributes to resistance in PDAC in mouse models and facilitates tumor immune evasion [282, 283].
PD-1 and its ligand PD-L1 have shown durable antitumor responses and may improve survival in cancers such as lung, kidney, melanoma, and bladder cancers [284]. However, resistance to ICIs therapy is common, and only a subset of patients benefit from ICIs [285]. Resistance to ICIs may be related to the inability of lymphocytes to penetrate the tumor, a phenomenon known as immune rejection, and is associated with increased ECM stiffness [286]. Combining ICIs with other therapies may improve outcomes, which include the combination of ICIs and chemotherapy, ICIs and radiotherapy, as well as ICIs and ICIs [287].
CAR-T cell therapy is an innovative type of immunotherapy. CAR is a surface protein consisting of a variety of sequentially arranged protein domains, enabling the specific detection of target antigens [255]. In CAR-T cell therapy, T cells are collected from the patient’s blood, genetically modified so that these T cells express CARs on their surface, cultured in vitro to expand their numbers, and then returned to the patient, thereby targeting and killing tumors in vivo [256, 257].
ECM is involved in guiding CAR T cells to infiltrate into the tumor. Due to the higher rigidity and fibrous matrix of the ECM, it can hinder CAR T cells from infiltrating into the center of the tumor core [288, 289]. The remodeling of the extracellular matrix is closely related to the therapeutic efficacy of CAR-T cell therapy. A patient’s T-cell condition is closely associated with previous treatment, the tumor burden, age, and disease, suggesting that the treatment may not be effective for all patients [290].
Although CAR T cells have shown some progress in hematologic malignancies, monotherapy is still ineffective in treating solid tumors [291]. The primary challenges for CAR-T cell therapy in solid tumors are the lack of appropriate tumor antigens and the inaccessibility of these antigens [292]. Additionally, the majority of solid tumors are prone to weakening CAR-T therapy efficiency by inducing immune checkpoint inhibitors (e.g., PD-1) [293]. Combining immune checkpoint blocking can synergistically amplify the effect of CAR-T cell therapy [258]. Thus, immune checkpoint blockers coupled with CAR-T cell therapy have the potential to improve treatment response and survival rates in patients with early-stage solid tumors [259]. Furthermore, multi-antigen targeting CAR-T cell therapy, such as hybrid, dual, or trivalent CAR T cells, can overcome tumor immune evasion and improve immunotherapy efficiency [260]. For instance, immunosuppressants or cytokine inhibitors combined with CAR-T therapy significantly improve the anti-tumor efficiency of hematological tumors [294].
Chimeric antigen receptor natural killer (CAR-NK) cells and chimeric antigen receptor macrophages (CAR-M) have emerged as promising alternatives to CAR-T cell therapies for the treatment of solid tumors. CAR-NK cells, which do not require an HLA match, possess the ability to target and eliminate tumor cells that lack major histocompatibility complex (MHC) molecules, thereby substantially reducing the risk of graft-versus-host disease (GvHD) and minimizing associated side effects. Furthermore, CAR-NK cells can be produced in large quantities from a variety of sources, highlighting their potential as off-the-shelf therapeutic products. The unique dual mechanisms of action—both CAR-dependent and non-CAR-dependent-provide NK cells with an added advantage in tumor eradication. These inherent benefits, particularly their reduced toxicity and the ability to be readily available as off-the-shelf products, make CAR-NK cells an attractive option for cancer immunotherapy. Nevertheless, CAR-NK therapies are not immune to challenges similar to those faced by CAR-T cell therapies, such as effective tumor site infiltration and overcoming immunosuppressive TME. A critical limitation of CAR-NK cell therapy lies in its limited in vivo persistence and the absence of sustained therapeutic effects, necessitating repeated high doses, which in turn exacerbates production costs [295].
CAR-M, on the other hand, offers distinct advantages over CAR-T and CAR-NK cells in addressing several key challenges inherent in solid tumor treatment, particularly regarding migration and infiltration into the immunosuppressive TME. Unlike CAR-T cells, which are derived from a single source, CAR-M cells can be harvested from a broad array of reliable sources, significantly reducing the risk of GvHD. This positions CAR-M as a promising candidate for allogeneic cellular immunotherapy in the context of solid tumors, with ongoing research exploring its phagocytic capabilities, tumor antigen presentation, and substantial tumor infiltration potential. CAR-M therapies provide a multifaceted approach to tumor destruction by leveraging both innate and adaptive immune response. However, the application of CAR-M faces a range of challenges, particularly in the form of cytokine release syndrome (CRS) and off-target toxicity, owing to the widespread distribution of macrophages in the body. This distribution may lead to unintended toxicity, which could undermine the therapeutic efficacy of CAR-M. Consequently, further investigation is imperative to refine CAR-M preparation techniques and ensure its clinical safety. Recent studies emphasize the importance of interactions between both modified and unmodified macrophages and T cells or NK cells in facilitating tumor regression, suggesting that combining CAR-M with CAR-NK or CAR-T cells may further enhance its antitumor activity [258].
Biophysical properties of the ECM determine tumor immune response. The biomechanical properties (e.g., stiffness and elasticity), topographical structure (e.g., fiber orientation and pore size), and biochemical properties (e.g., differences in ECM composition and cytokine concentration distribution) collectively regulate immune-cells motility, penetration, and activity, thus influencing tumor immune evasion and therapeutic response [296, 297]. Previous research has shown that the abnormal biomechanical and chemical characteristics of the ECM not only hinder immune-cell infiltration and function but also promote cancer resistance to immunotherapy through mechano- and chemical-signal transmission processes [16]. Therefore, understanding ECM biomechanics and ECM–immune cell coupling mechanisms is crucial for elucidating immune evasion and guiding the development of new therapies. Future research should focus on: (1) Modifying ECM mechanical properties (stiffness, flexibility) and surface topography to optimize immune-cell infiltration and activity, thereby enhancing immunotherapy effectiveness. Investigations into molecules or pathways responsive to ECM stiffness, such as integrin-mediated adhesion and immune-cell migration, could inform the design of targeted drugs or biomaterials that soften the ECM. Altering the ECM surface to present additional adhesion molecules or cytokines could also enhance immune cell binding and activation. For example, engineering the ECM surface to display more adhesion molecules or cytokines may attract immune cells and improve therapy; (2) Elucidating the molecular mechanisms of ECM–immune cell coupling: further study of how ECM regulates immune cells through integrins, mechanosensitive ion channels, and other mediators can provide guidance for targeted treatment. Exploring crosstalk between integrin signaling and pathways such as MAPK and PI3K–Akt could yield deeper insights, for instance, how integrin activation in T cells influences proliferation and cytokine production. The role of mechanosensitive ion channels (e.g., Piezo, TRPV) in various immune-cell types warrants further investigation. Understanding how mechanical signals transmitted via these channels regulate gene transcription involved in immune-cell differentiation may reveal new targets to manipulate immune cell fate and boost anti-tumor immunity; (3) Combining ECM-targeted therapy with immunotherapy: new treatments can be developed based on ECM biomechanical properties, including enzymes that degrade ECM and nanomedicines targeting ECM degradation. For example, nanoparticles loaded with MMP inhibitors could be designed to release only in response to tumor markers, ensuring localized ECM degradation and enhanced penetration of immunotherapies such as immune checkpoint inhibitors and CAR-T cells. Designing nanoparticles with high affinity for ECM components (e.g., collagen, hyaluronic acid) enables targeted delivery of drugs, siRNAs, or immune-stimulating molecules to the tumor microenvironment. Nanoparticles that co-deliver immune checkpoint inhibitors and ECM-targeting agents may further enhance tumor accumulation and immune response. Preclinical and clinical evaluations should assess the efficacy of this nanomedicine-based combination therapy for overcoming resistance. Combining ECM-degrading enzymes with current immunotherapies (such as ICIs and CAR-T cell therapy) may yield synergistic effects: ECM degradation can expose more tumor antigens, resulting in improving immune access, while also disrupting immunosuppressive microenvironments created by ECM. Future studies could determine optimal timing and dosing for combining ECM-degradation enzymes with various immunotherapies (e.g., sequencing of MMP inhibitors and anti-PD-1 antibodies) to maximize anti-tumor effects.
In recent years, artificial intelligence (AI) and machine learning (ML) have found broad applications in biomedical research, offering promising avenues for the development of ECM-targeted immunotherapies. AutoML significantly enhances the efficiency of integrating multimodal ECM data—such as imaging, genomics, and clinical information-by automating data preprocessing, feature engineering, and model tuning. In the context of clinical prognosis prediction, AutoML frameworks like AutoGluon and TPOT have outperformed conventional ML models, exhibiting superior AUROC performance. These frameworks are particularly suited to exploring the correlation between ECM protein expression profiles and patient survival data. Moreover, AutoML has shown distinct advantages in genomic sequence analysis, as exemplified by the deepRAM model, which employs a hybrid architecture combining Convolutional Neural Networks (CNNs) and Recurrent Neural Networks (RNNs), optimized through random search and cross-validation. This approach enables efficient identification of protein-binding sites within DNA/RNA sequences, while simultaneously enhancing biological interpretability through the extraction of sequence motifs. AI-driven technologies facilitate precision transformation strategies by integrating mechanical signals from the ECM, immune-cell phenotypes, and genomic data [298, 299]. However, the application of AI in ECM research is currently hindered by a critical bottleneck: although interpreting feature importance in existing machine learning models often surpasses traditional methods, the biological interpretation of these features still requires dedicated signal-pathway analyses for confirmation. Moreover, current ECM models are based on static data, which does not effectively capture the dynamic characteristics of the tumor ECM. Looking ahead, the development of multi-scale AI models that integrate both physical mechanics and biochemical signals is imperative. The fusion of these models with interpretable AI (XAI) technologies holds the potential to shift the focus from mere correlation analysis to causal mechanistic understanding. Furthermore, the application of transformer architectures to handle spatiotemporal sequence data of ECM-immune cell interactions is poised to unravel the dynamic regulatory patterns governing the heterogeneity of the TME. These advances could provide a robust theoretical foundation for the development of next-generation ECM-targeted immunotherapies.
ECM has become a vital constituent in the TME, affecting the growth and development of cancer. Because of low side effects and toxicity, immunotherapy has opened the door for cancer therapy while eliminating the drawbacks of conventional tumor therapies. However, due to various factors, including cancer heterogeneity, rejection, and infiltration of immune cells, immunotherapy is often unable to work as well as expected. With a deeper understanding of the correspondence between the biophysical properties of the tumor ECM and the tumor immune response, and by continuing to develop ECM-targeted immunotherapy, new advances in cancer immunotherapy are expected. These advancements could lead to more effective treatments and substantial improvements in outcomes for cancer patients.
XZ, FK, DW and QL conceptualized the insights. YL, XYL, XXL, JZ, XG, CS and CC performed literature research and analyzed the data. MY and HY drew figures. XZ wrote the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This work was financially supported by The Science and Technology Strategic Cooperation Programs of Luzhou Municipal People’s Government and Southwest Medical University (2024LZXNYDJ074, 2024LZXNYDJ055), Southwest Medical University (2024ZKZ008, 05/00170050), and the Undergraduate Training Program for Innovation and Entrepreneurship (2025314, 2025286, 202410632054, 2024303).
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.