
- Academic Editor
Research on the molecular progression of esophageal squamous dysplasia to cancer remains limited. The majority of prior studies have focused on morphological precancerous lesions sampled adjacent to tumors, and have relied primarily on the analysis of data from whole-exome sequencing.
To investigate the development of esophageal squamous cell carcinoma (ESCC), whole genome analysis was conducted on 13 precancerous tissues and 15 ESCC tissues. Field effects were avoided by using biopsies of squamous dysplasia from patients without concurrent tumor, thereby allowing study of molecular alterations associated with the true precancerous state.
Our results revealed frequent copy number alterations (CNAs) and structural variants (SVs) in esophageal squamous dysplasia. These changes were also detected in ESCC, indicating that genomic instability markers such as CNAs and SVs occur at an early stage and persist throughout ESCC evolution. The detection of TP53 mutations and CASP8 deletions in both premalignant lesions and ESCC suggests they may be early driving events during esophageal carcinogenesis. Mutations in MUC5B were observed in 7.7% of precancerous lesions and 6.7% of ESCC. Moreover, these mutations were associated with a higher tumor mutational burden (TMB) and an immune “hot” tumor microenvironment. Apolipoprotein B mRNA-editing catalytic polypeptide-like (APOBEC) enzyme-associated mutational signatures were exclusively identified in ESCC and may further exacerbate genomic instability in the more advanced stages of tumorigenesis. Significantly higher ploidy alterations levels were detected in ESCC compared to squamous dysplasia. Moreover, the cohort that underwent local recurrence of dysplasia within two years had significantly elevated ploidy alterations levels compared to those with no long-term recurrence. These results indicate that elevated levels of aneuploidy and genomic instability were associated with tumor progression and local recurrence of dysplasia.
Mutations in TP53 and MUC5B, as well as deletion of CASP8, may be early driver events in carcinogenesis and could precede the emergence of the APOBEC mutation signature. Moreover, ploidy alterations confer a selective advantage to genomically unstable cells, thereby promoting their progression toward malignant transformation. Collectively, our results demonstrate that genomic instability is prevalent in precancerous lesions and intensifies during the late stages of tumor progression. Cells with a certain level of genomic instability appear to possess a competitive advantage for malignant transformation.
Esophageal carcinoma ranks as the seventh most prevalent malignancy and the sixth leading cause of cancer-related mortality worldwide, accounting for over 540,000 annual deaths globally [1]. The two major pathological subtypes of esophageal cancer are esophageal squamous cell carcinoma (ESCC), and esophageal adenocarcinoma (EAC). They exhibit marked differences in epidemiology, molecular profiles, and oncogenic pathways [2]. ESCC is the predominant subtype in Asian populations, whereas EAC is more common in Western countries [3, 4]. ESCC is considered to be more analogous to other types of squamous cell carcinoma, whereas EAC exhibits notable similarities to gastric cancer [5]. Despite these differences, both subtypes follow a progressive carcinogenic sequence from precursor lesions to invasive carcinoma. The progression from normal epithelium to precursor lesions, and ultimately to invasive cancer, provides an invaluable model for studying the full trajectory of esophageal tumorigenesis. Uncovering the molecular mechanisms driving the transition from pre-invasive to invasive disease is critical for improving early detection and therapeutic strategies [6]. Over the past decade, significant progress has been made in understanding the mechanisms underlying the progression of Barrett’s esophagus (BE), a metaplastic precursor lesion to EAC. This has resulted in significant advances in surveillance protocols of EAC [7, 8, 9, 10, 11]. However, in contrast to EAC, research on esophageal squamous dysplasia and its progression to ESCC remains limited, leaving significant gaps in our understanding of this pathway.
Although several studies have compared the genomic sequencing data of ESCC and adjacent precancerous lesions, many critical questions remain unanswered, particularly regarding molecular differences between the two histopathological stages. Liu Xi et al. [12] reported that dysplastic lesions adjacent to tumors exhibit similar alterations to ESCC in terms of both mutations and copy number alterations (CNAs). Chen Xixi et al. [13] found that “two-hit” events on TP53 are rare in dysplasia samples from tumor-free patients, in contrast to dysplastic lesions that are adjacent to tumors. Chang Jiang et al. [14] inferred that TP53 inactivation was one of the earliest steps in the malignant transformation of esophageal epithelium.
Most of the above studies focused on morphological precancerous lesions that were adjacent to tumors, and relied primarily on data from whole-exome sequencing (WES) for their analysis. However, it is important to note that due to field cancerization, dysplastic lesions adjacent to tumors are more likely to represent the final stages of cancer evolution. Consequently, it is not known to what extent these lesions accurately reflect earlier time points in the natural history of neoplastic transformation [7]. For instance, genetic alterations observed in precancerous lesions adjacent to tumors may differ from those found in precancerous lesions in patients who have not progressed to cancer [15]. Furthermore, many insights into neoplastic transformation can only be obtained through whole-genome analysis, and would not be seen with targeted exome sequencing [16]. Currently, there are few reports on the state of structural variants (SVs) in precursors to ESCC.
To address these gaps, we conducted whole-genome analysis on precancerous samples from patients without concurrent cancer, as well as on ESCC samples. By analyzing the genomic landscape of esophageal premalignant lesions, we detected molecular similarities and differences between dysplasia and cancer. Apart from finding some well-known CNAs known to act as drivers, our findings also revealed that SVs occur frequently in esophageal squamous dysplasia, indicating they emerge early in the evolution of ESCC. Moreover, our data showed that the level of ploidy alterations could distinguish esophageal squamous dysplasia from ESCC, and were also associated with the local recurrence of dysplasia. Based on these findings, we propose that elevated levels of aneuploidy occur relatively late in the progression of ESCC and exacerbate genomic instability, thereby providing cells with a competitive advantage for malignant transformation.
Two cohorts of fresh samples were obtained: pairs of dysplastic and normal samples from 13 patients without ESCC, and pairs of tumor and normal samples from 15 ESCC patients. None of the patients included in the study had received any treatment prior to surgery or biopsy. Sample collection was conducted following approval from the Institutional Review Board of the Cancer Hospital, Chinese Academy of Medical Sciences (Ethic Approval Number: 12-71/605). The study was carried out in accordance with the guidelines of the Declaration of Helsinki. A written consent was signed by the patients or their families/legal guardians.
Whole-genome sequencing was performed on all samples using the Illumina NOVA sequencing platform. The dataset included 13 dysplastic samples paired with morphologically normal esophageal epithelium, and 15 tumor samples paired with morphologically normal esophageal epithelium. The median sequencing depth achieved was 8.8X.
To identify ESCC driver genes, we curated a comprehensive gene set by combining 200 pan-cancer and esophageal cancer driver genes reported in the literature [17] with 98 esophageal cancer driver genes downloaded from the IntOGen database [18]. This yielded a total of 253 ESCC-related driver genes for subsequent analysis.
Strelka software (version 2.9.10; Illumina, San Diego, CA, USA) was used to detect somatic mutations in the sequencing data. Biological hallmarks enrichment analyses were performed using the Metascape database. The facets tool (version 0.6.0) was used to analyze CNAs. We employed ichorCNA (version 2.0) to calculate ploidy alterations for each sample. Meerkat algorithm was used to detect SVs. GeneFuse [19] (version 0.6.1) was applied to detect target gene fusions by scanning FASTQ files.
Expression data,genomic data and associated clinical information for ESCC
samples were obtained from the cancer genome atlas (TCGA) database. After
retaining samples that included both sequencing data and clinical information, a
total of 82 samples were selected for subsequent analysis. The R package
“immunedeconv” and EPIC algorithm were employed to ensure accurate assessment
of immune scoring. The Limma package in R software (version 4.0.3; R Foundation for Statistical Computing, Vienna, Austria) was used to
study the differentially expressed genes. We defined “p
Comparisons between the two groups mentioned in the study was performed using
the Wilcoxon rank-sum test. A two-sided p-value
This study enrolled 28 patients, consisting of 13 cases of pathologically
confirmed and tumor-free esophageal squamous dysplasia, and 15 cases of
early-stage ESCC. The clinical characteristics of the two cohorts are summarized
in Tables 1,2, respectively. The dysplasia cohort comprised predominantly
high-grade intraepithelial neoplasia (84.62%, 11/13), with 23.08% (3/13)
exhibiting dual smoking/alcohol exposure. The ESCC cohort consisted of 8 cases
with tumor-nodes-metastasis (TNM) stage I disease and 7 with TNM stage II
disease. These samples represent relatively early-stage cancer and are closer to
precursor lesions in terms of evolution, thereby providing a temporally
compressed model to identify driver events that orchestrate the transition from
intraepithelial neoplasia to early invasive carcinoma. Moreover, for the purposes
of risk stratification, we conducted a prospective follow-up of the precancerous
lesion cohort for
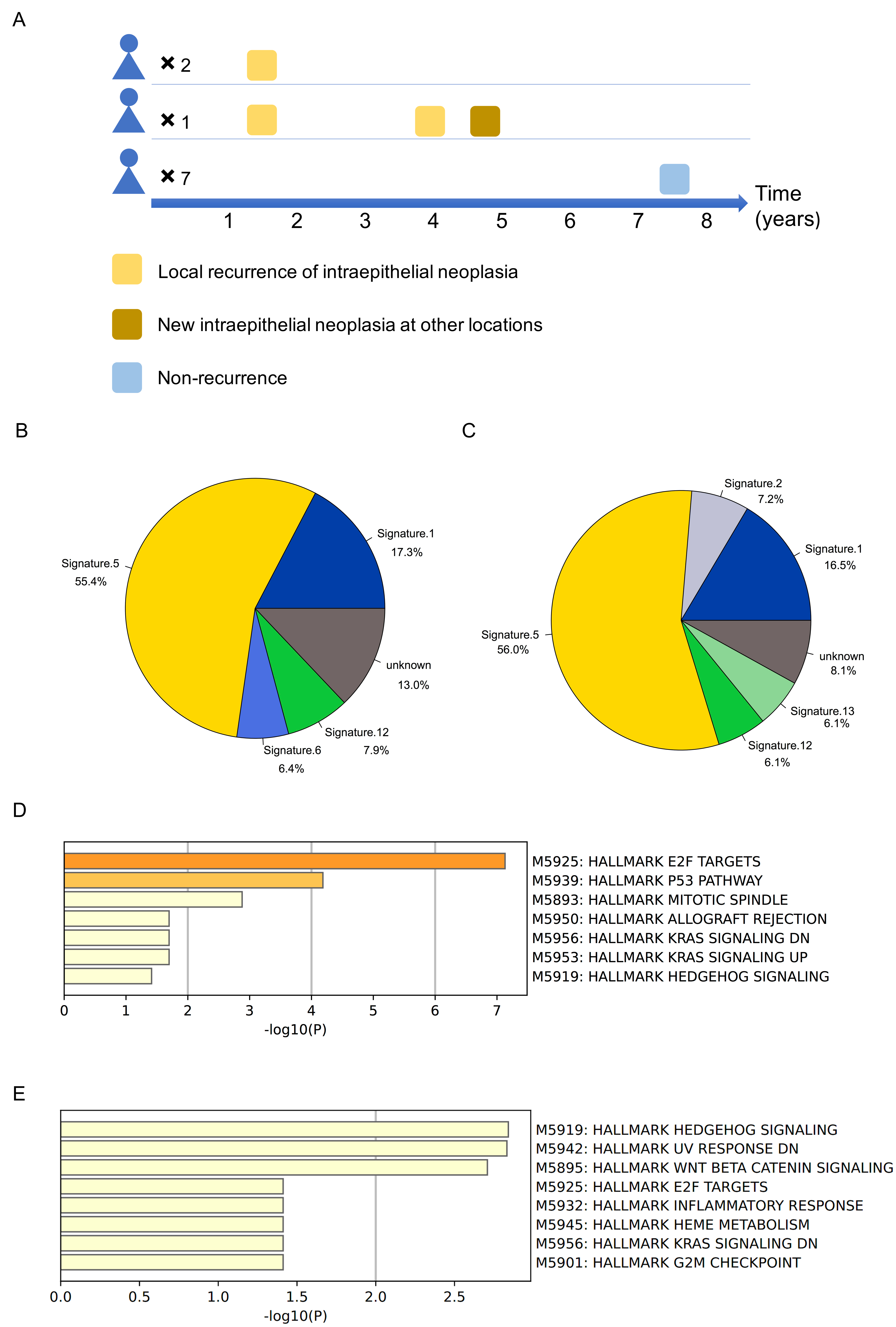 Fig. 1.
Fig. 1.
Follow-up outcomes in the dysplasia group and mutation signatures, hallmarks associated with genes harboring nonsynonymous mutations in the two groups. (A) Follow-up outcomes in the dysplasia group. (B) Mutation signatures in the dysplasia group. (C) Mutation signatures in the ESCC group. (D) Hallmarks associated with genes harboring nonsynonymous mutations in the dysplasia group. (E) Hallmarks associated with genes harboring nonsynonymous mutations in the ESCC group. ESCC, esophageal squamous cell carcinoma.
| Clinical characteristic | Number of patients (%) | |
| Gender | ||
| Male | 7 (53.85%) | |
| Female | 6 (46.15%) | |
| Grade | ||
| Low-grade intraepithelial neoplasia | 2 (15.38%) | |
| High-grade intraepithelial neoplasia | 11 (84.62%) | |
| Smoking history | ||
| Yes | 3 (23.08%) | |
| No | 10 (76.92%) | |
| Drinking history | ||
| Yes | 3 (23.08%) | |
| No | 10 (76.92%) | |
| Post-treatment follow-up | ||
| Relapse of intraepithelial neoplasia | 3 (23.08%) | |
| No relapse | 7 (53.85%) | |
| Lost to follow-up | 3 (23.08%) | |
| Total | 13 | |
Due to rounding, percentages may not sum to 100%.
| Clinical characteristic | Number of patients (%) | |
| Gender | ||
| Male | 11 (73.33%) | |
| Female | 4 (26.67%) | |
| T stage | ||
| T1 | 11 (73.33%) | |
| T2 | 1 (6.67%) | |
| T3 | 3 (20.00%) | |
| N stage | ||
| N0 | 12 (80.00%) | |
| N1 | 3 (20.00%) | |
| M stage | ||
| M0 | 15 (100%) | |
| TNM stage | ||
| I | 8 (53.33%) | |
| II | 7 (46.67%) | |
| Smoking history | ||
| Yes | 7 (46.67%) | |
| No | 8 (53.33%) | |
| Drinking history | ||
| Yes | 7 (46.67%) | |
| No | 8 (53.33%) | |
| Total | 15 | |
ESCC, esophageal squamous cell carcinoma; TNM, tumor-nodes-metastasis.
The non-negative matrix factorization (NMF) method was used to identify
mutational signatures. This revealed the squamous dysplasia and ESCC groups
exhibited a similar mutational context (Fig. 1B,C). Notably, both groups showed a
strong enrichment of Signature 5. According to the Catalogue of Somatic Mutations
in Cancer (COSMIC) database, Signature 5 exhibits transcriptional strand bias for
T
We adopted strict screening criteria to ensure the authenticity of the mutation
results. Through stringent filtering standards, we identified 41 nonsynonymous
single nucleotide variants (SNVs) in the dysplasia group and 53 in the ESCC
group. Among these, TP53 demonstrated the highest
mutation frequency, occurring in 54% (7/13) of squamous dysplasia samples and
27% (4/15) of ESCC samples. This indicates that it serves as an early initiating
event in the carcinogenesis of ESCC. Four genes (TP53, NOTCH1,
MUC5B, and MLL2) were found to be mutated in
both cohorts, suggesting they represent early events in the progression of ESCC.
MLL2, an essential histone regulator gene, is frequently altered in ESCC
[24]. MUC5B encodes structurally related mucin glycoproteins [25] and is
known to facilitate cell-to-cell and cell-to-matrix interactions, as well as
cell-autonomous signaling, thus promoting tumorigenesis and distant dissemination
of tumor cells [26]. However, MUC5B is rarely reported in the
mutational spectrum of cancer, suggesting it may represent a novel driver gene in
ESCC progression. To further explore the role of MUC5B, we
analyzed sequencing data from ESCC, head and neck squamous cell carcinoma
(HNSCC), and lung squamous cell carcinoma (LSCC) obtained from the cBio Cancer
Genomics Portal [27, 28]. The mutation frequencies for MUC5B were 4% in
ESCC, 7% in HNSCC, and 13% in LSCC (Fig. 2A and Supplementary Fig.
1A,B). Additionally, MUC5B mutation was correlated with higher
tumor mutational burden (TMB) (nonsynonymous) in ESCC, HNSCC and LSCC (Wilcoxon
test; p = 0.007, p = 8.67
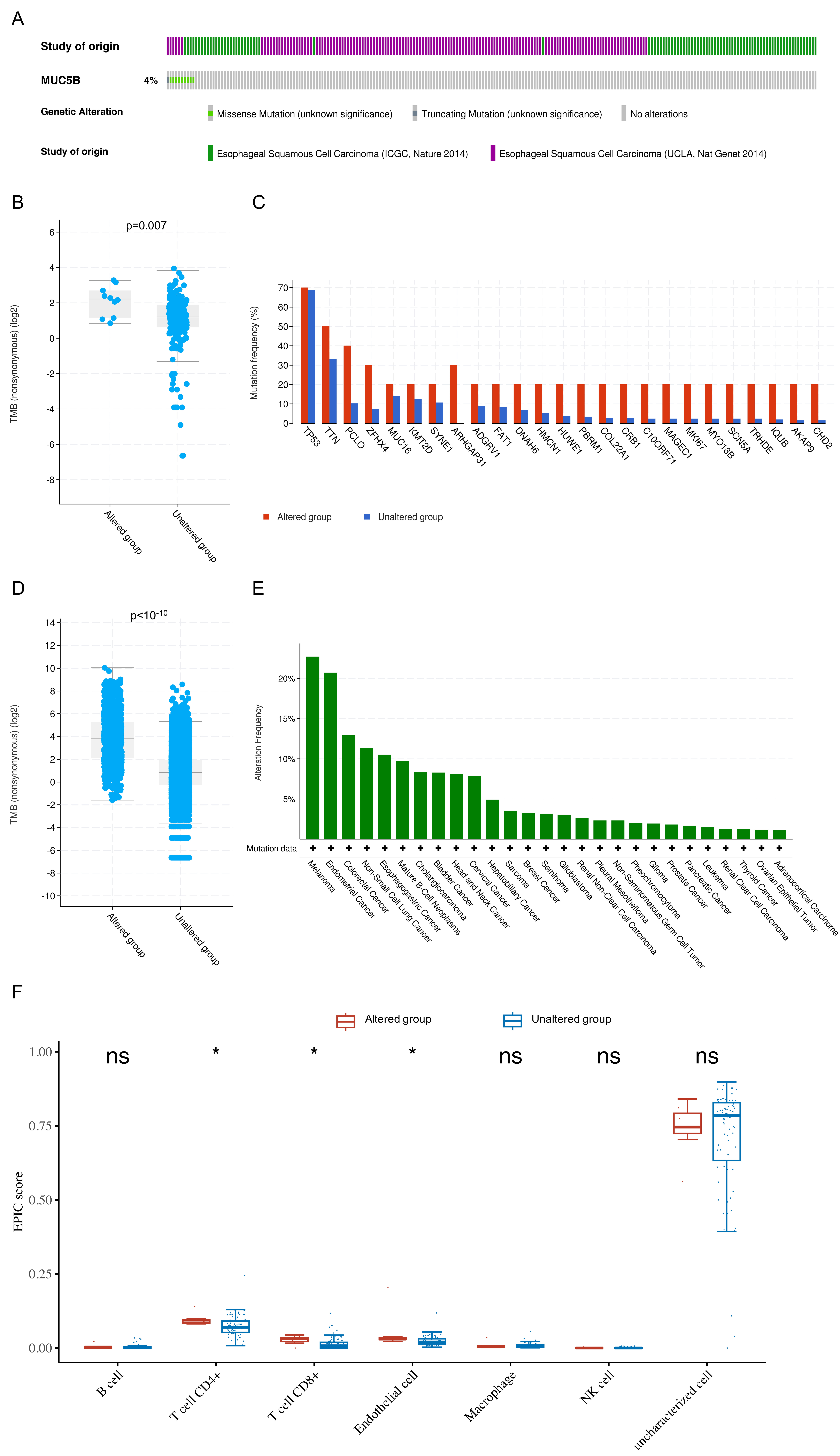 Fig. 2.
Fig. 2.
Mutation frequency of MUC5B in ESCC and pan-cancer
datasets, the relationship between MUC5B mutations and TMB and the
relationship between MUC5B mutations and tumor-infiltrating immune
cells. (A) Mutation frequency of MUC5B in ESCC datasets from the cBio
Cancer Genomics Portal. (B) Comparison of TMB between MUC5B-mutated and
MUC5B wild-type groups in ESCC datasets from the cBio Cancer Genomics
Portal. (C) Comparison of the mutation frequency of recurrently mutated genes
between MUC5B-mutated and MUC5B wild-type cohorts in ESCC
datasets from the cBio Cancer Genomics Portal. (D) Comparison of TMB between
MUC5B-mutated and MUC5B wild-type groups in pan-cancer datasets
from the cBio Cancer Genomics Portal. (E) Mutation frequency of MUC5B in
pan-cancer datasets from the cBio Cancer Genomics Portal. (F) Comparison of
tumor-infiltrating immune cells between MUC5B-mutated and wild-type
groups in ESCC datasets from TCGA. * p
Since TMB is used as a biomarker for tumor immunotherapy, we conducted an in-depth analysis of the relationship between MUC5B mutations and tumor-infiltrating immune cells in ESCC using the EPIC algorithm and the sequencing data in TCGA. The MUC5B-mutated group exhibited significantly higher infiltration levels of CD4+ T cells, CD8+ T cells, and endothelial cells compared to the MUC5B wild-type group (Wilcoxon test, p = 0.026, p = 0.022, p = 0.027, respectively) (Fig. 2F). This suggests the MUC5B-mutated group is characterized by an immune “hot” tumor microenvironment. To further explore the functional implications of MUC5B mutations, we analyzed the expression profile differences between mutant and wild-type groups, finding 252 upregulated and 134 downregulated genes in the MUC5B-mutated group. Enrichment analyses showed upregulated genes were mainly linked to phospholipid metabolism, cell adhesion, and platinum drug resistance (Supplementary Fig. 2A,B), while downregulated genes were associated with chromatin assembly pathway and rheumatic and autoimmune disease pathway (Supplementary Fig. 2C,D). Downregulation of genes enriched in chromatin assembly pathway suggests that core processes governing nucleosome formation and chromatin organization are attenuated in the MUC5B-mutant group, resulting in elevated genomic instability. Subsequently, we conducted GSEA analysis to characterize the KEGG pathways affected by the transcriptome change with the MUC5B mutation. We found 55 positively enriched pathways and 26 negatively enriched pathways with a q-value of less than 0.05. GSEA analysis further confirmed significant depletion of genome stability related pathways in the MUC5B-mutated group, including DNA replication, mismatch repair, homologous recombination, and base excision repair (Supplementary Fig. 3).
Next, we analyzed the biological hallmarks of genes harboring nonsynonymous mutations. Those harboring nonsynonymous mutations in the dysplasia group were mainly clustered in the hallmarks for E2F targets, TP53 pathway, mitotic spindle, and KRAS and HEDGEHOG signaling (Fig. 1D). In addition to the cell cycle, KRAS, and HEDGEHOGsignaling hallmarks observed in the precancerous group, nonsynonymous mutated genes in the ESCC group were also involved in WNT and inflammatory response hallmarks (Fig. 1E).
We next conducted CNA analysis to better decipher the genomic alterations during esophageal carcinogenesis. The median length of genome affected by CNAs in precancerous cohorts was 20.52 MB, including both gain and loss of copy number, while that of the ESCC cohort was 23.45 MB. Although the level of median copy number alteration was higher in ESCC than in esophageal squamous precursor, this was not significantly different (Wilcoxon test, p = 0.82) (Fig. 3A). Loss of heterogeneity (LOH) is ubiquitous in cancer, and studies have shown that LOH can contribute to the inactivation of some tumor suppressor genes, thereby affecting the occurrence and progression of cancer. We found that esophageal precancerous lesions and ESCC shared a similar LOH burden (Wilcoxon test, p = 0.16) (Fig. 3B), suggesting that LOH already occurs frequently in precancerous lesions.
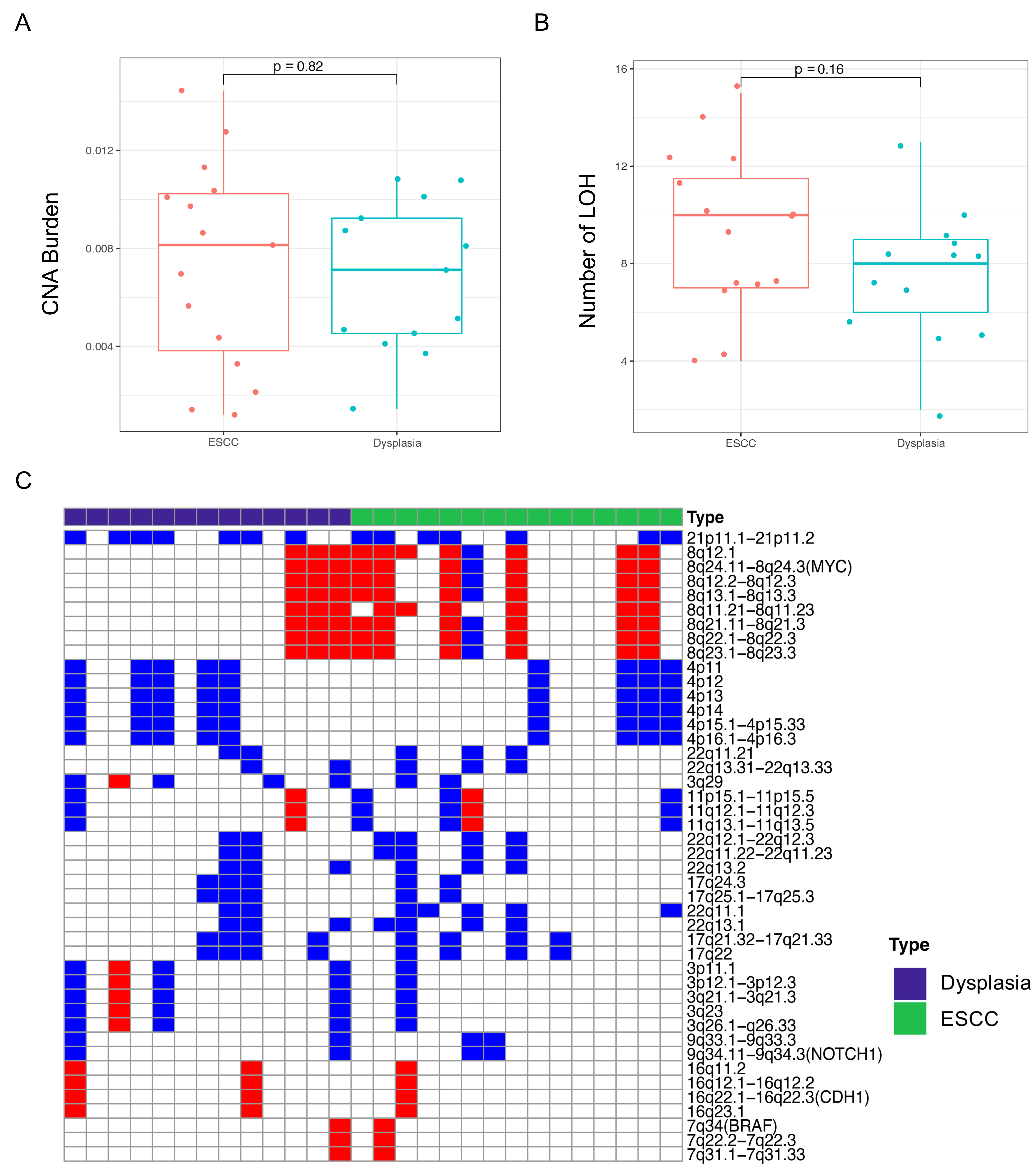 Fig. 3.
Fig. 3.
CNA profiling across the dysplasia and ESCC groups. (A) Comparison of the CNA burden between ESCC and dysplasia samples. (B) Comparison of the number of LOH events between dysplasia and ESCC samples. (C) Frequently amplified and deleted segmented CNAs in the dysplasia and ESCC groups. Red indicates copy number amplification, and blue indicates copy number deletion. CNA, copy number alteration; LOH, loss of heterogeneity.
We also analyzed segmented CNAs that were frequently amplified or deleted. Recurrent CNA profiles demonstrated both overlapping and divergent patterns between premalignant and malignant lesions (Fig. 3C). Focal CNA analysis identified 8q amplification as a shared event in both premalignant lesions and malignant tissues. This segment, reported as a hotspot in ESCC, contains the cancer-related gene MYC, which plays a critical role in regulating cell proliferation. These findings suggest that amplification of the 8q segment may be a key driving event in carcinogenesis. In addition, we identified several alterations in chromosomal segments in esophageal squamous precancerous lesions that have been rarely reported in previous studies. For instance, a 7q segment containing the driver gene BRAF was found to be amplified in precancerous lesions. BRAF is involved in regulation of the MAPK/ERK signaling pathway, which affects cell division, differentiation and secretion. Moreover, we identified frequent deletions that affect segments in 4p, 9q (containing the driver gene NOTCH1), and 17q. The deletion in 4p has been frequently reported in ESCC [29], while LOH of 17q has also been identified in ESCC [30].
SVs play a mysterious role in cancer through their ability to move blocks of adjacent genes simultaneously, or to generate gene fusion, thereby contributing to concurrent oncogenic events [31, 32]. However, the landscape of SVs in esophageal squamous dysplasia remains unclear. To address this, we conducted SV analysis through the Meerkat computational tool [33, 34]. A median of 6300 SVs per genome (range 3684–8308) were identified in the dysplasia cohort, and 3616 (range 2751–5184) in the ESCC cohort, with no statistically significant difference between the two (Wilcoxon test, p = 0.41). Five SV types were detected, consisting of duplications/intra-chromosomal translocations, deletions/intra-chromosomal translocations, inversions and two types of inter-chromosomal translocations (type 1 and type 2) [34] (Fig. 4A). Genome-wide SV analysis revealed that inter-chromosomal translocation was the predominant SV type in both precancerous and cancerous tissues.
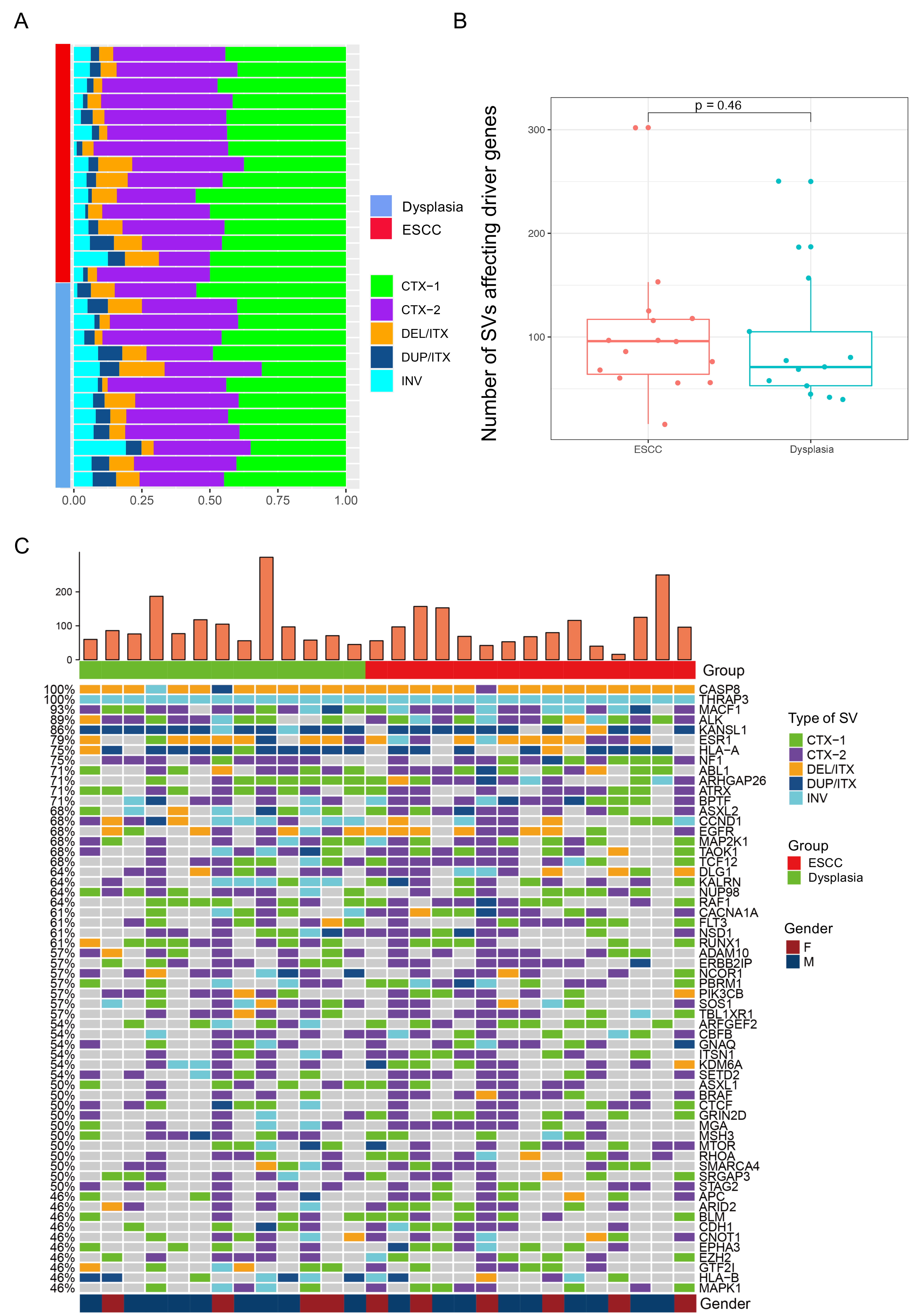 Fig. 4.
Fig. 4.
SV profiling across the dysplasia and ESCC groups. (A) The proportions of different SV types in each sample. (B) Comparison of the number of SVs affecting driver genes between dysplasia and ESCC samples. (C) Landscape of SVs affecting driver genes in dysplasia and ESCC samples. SV, structural variant; CTX, inter-chromosomal translocation; ITX, intra-chromosomal translocation; DEL, deletion; DUP, duplication; INV, inversion; F, female; M, male.
To further study the potential impact of the large number of SVs on biological processes, these were mapped at the gene level and the frequently rearranged driver genes in esophageal cancer were analyzed. No significant difference in the frequency of SVs that disrupt ESCC driver genes was observed between the dysplasia and tumor cohorts (Wilcoxon test, p = 0.46) (Fig. 4B).
We next analyzed and characterized the spectrum of driver genes with a high frequency of SVs in both groups (Fig. 4C). CASP8, a recently identified novel driver gene associated with ESCC [35], exhibited the highest frequency of SVs in both the dysplasia and tumor groups, predominantly in the form of deletions. While deletions in CASP8 have been reported in ESCC [36], the involvement of CASP8 in the precursor stage of esophageal lesions has not been previously documented. CASP8, a key initiator of death receptor-mediated apoptosis, also plays a critical role in suppressing RIPK3-MLKL-dependent necroptosis [37, 38]. This gene is essential for preventing tissue damage during both embryonic development and adulthood [37], and its deletion can lead to ileocolitis and gut barrier dysfunction [39]. KANSL1 was also frequently rearranged in both cohorts. Amplifications and rearrangements of KANSL1 were previously reported in ovarian cancer, and it could serve as a potential biomarker and therapeutic target for immune response modulation [40]. KANSL1 was also found to be enriched in tumors with genome doubling events [41]. ESR1 is frequently affected by deletion-type SVs in both precancerous lesions and tumor samples. High frequencies of ESR1 SVs were reported in inflammatory reflux esophagitis, Barrett’s metaplasia, dysplasia, and EAC [42].
SVs have been shown to mediate oncogenic gene fusion events. The present analysis identified 169 fusion genes that were shared between premalignant and malignant esophageal lesions. Among these, the kinase-associated SND1-BRAF fusion, previously reported in thyroid carcinomas [43], was detected in both premalignant (1/13, 7.7%) and malignant (1/15, 6.7%) samples.
Ploidy alterations were also compared between the dysplasia and cancer groups. The average ploidy was found to be 2.025 in precursor lesions and 2.31 in the ESCC group (Fig. 5A), demonstrating a significantly higher degree of genomic instability in the cancer group (Wilcoxon test, p = 0.029). Importantly, the cohort that underwent local recurrence of dysplasia within two years had significantly elevated level of ploidy alterations compared to those with no long-term recurrence (Wilcoxon test, p = 0.033). This finding suggests that higher level of ploidy alterations may serve as a predictive marker for an increased risk of recurrence (Fig. 5B).
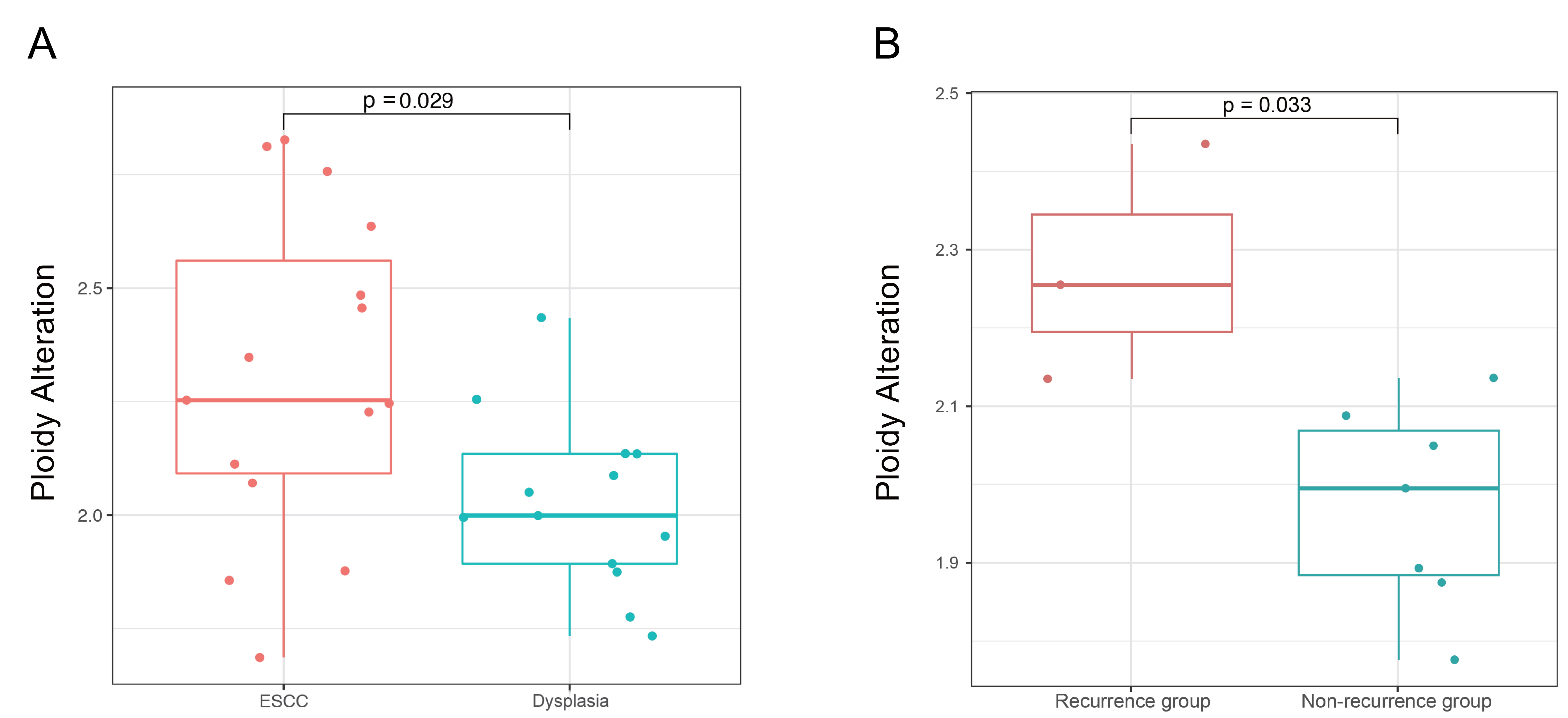 Fig. 5.
Fig. 5.
Comparison of ploidy alterations between dysplasia and ESCC samples and comparison of ploidy alterations between recurrence group and non-recurrence group. (A) Comparison of ploidy alterations between dysplasia and ESCC samples. (B) Comparison of ploidy alterations between recurrence group and non-recurrence group in the dysplasia cohort.
The current study comprehensively analyzed the whole genomic landscape of esophageal squamous precancerous lesions and ESCC. Field effects, which encompass tumor-host interactions and gene-environment interactions [44], can remodel the neighboring microenvironment of tumor cells. Tumorous dysplasia exhibits a more malignant molecular phenotype compared to non-tumorous dysplasia, with a similar pattern and burden of genomic alteration to that of tumor tissue [12]. Such tumor-adjacent precancerous lesions may already have acquired some transformative potential [13]. Once the cancerous tissue is removed and suppression of the dominant ESCC clone is lifted, the lesion may rapidly progress into carcinoma. However, among precursor cases from patients with no sign of ESCC, 33% progressed or recurred, while others regressed or remained static [45]. To reduce the effects of field cancerization and to conduct risk stratification, we analyzed biopsies of squamous dysplasia obtained from patients without concurrent tumor, as well as performing long-term follow-up of these patients.
Our analysis revealed that precursor lesions without tumors harbor frequent CNAs and SVs, which differs from some previous reports on esophageal carcinogenesis. Our use of whole-genome sequencing generates a more comprehensive genomic profile compared to targeted or exome sequencing approaches, potentially explaining the discrepancy with earlier studies. The inherent heterogeneity of dysplasia may also contribute to the differences observed between studies. Despite the comparable burden of CNAs and SVs between precancerous lesions and ESCC, the overall genomic alteration burden was generally higher in ESCC. This result suggests that chromosomal instability, a hallmark of cancer, progressively accumulates during carcinogenesis. Similar to our finding, a large burden of canonical CNAs were also detected in the hyperplasia/metaplasia stages of the LSCC progression model, with their frequency also increasing during the progression of lesions [46].
Our results showed a high mutation frequency of TP53 in precancerous lesions. Chang Jiang et al. [14] reported that the frequency of TP53 biallelic inactivation increases dramatically in early precancerous lesion stage and TP53 inactivation leads to CNAs in TP53 biallelic loss clones. These results suggested that TP53 may be an early initiator of carcinogenesis. Additionally, we found the MUC5B gene was mutated in 7.7% of precancerous lesions and 6.7% of ESCC. This gene is implicated in cell-to-cell and cell-to-matrix interactions. Interestingly, mutations in MUC5B have been associated with increased TMB in ESCC, HNSCC, LSCC, and pan-cancer datasets, suggesting they may represent a novel early event in carcinogenesis and a biomarker of genomic instability. Moreover, our analysis revealed that samples with MUC5B mutations were associated with immunologically “hot” tumors, indicating a potentially heightened responsiveness to immunotherapy. Functional enrichment analysis of differentially expressed genes indicates that the MUC5B-mutant group is more prone to invasive metastasis and platinum-based chemoresistance and exhibits increased genomic instability owing to attenuated chromatin assembly. Due to the deficiency in homologous recombination function in the MUC5B-mutant group, these patients may derive therapeutic benefit from polyadenosine-diphosphate-ribose polymerase (PARP) inhibitors. Meanwhile, DNA-repair defects in the mismatch repair pathway within the MUC5B-mutant cohort are also biomarkers for guiding the use of immune-checkpoint inhibitors [47, 48]. Concurrently, individuals harboring MUC5B mutations may be more sensitive to novel treatment agents that target genomic instability, including KIF18A inhibitors, p53-reactivating agents and PLK4 inhibitors, all of which are presently being tested in clinical trials. Furthermore, the APOBEC mutagenesis signature, a known contributor to genomic instability, was detected exclusively in ESCC but was absent in precancerous lesions. This observation indicates the APOBEC mutagenesis pattern may represent a relatively late event in the carcinogenic process, potentially conferring invasive capabilities to neoplastic cells.
The recent advances in genomic research have focused increasing attention towards the study of genomic alterations in morphologically normal cells. Li Ruoyan et al. [49] reported that normal esophageal tissues already harbor CNAs. These are predominantly characterized by whole-chromosomal amplifications of chromosomes 3, 5, and 7. The present study also detected amplification of 7q in both dysplasia and ESCC, suggesting that gain of 7q may represent an early genomic event and play a role in the accumulation of genomic instability during the progression to ESCC.
SVs are well-known hallmarks of cancer, affecting a larger fraction of the genome than point mutations by increasing or deleting gene copy numbers. These frequent genomic rearrangements can result in the amplification of oncogenes or the disruption of tumor suppressor genes, thus contributing to tumorigenesis. In our study, CASP8, a recently identified driver gene, exhibited frequent deletions in premalignant lesions, indicating that SVs of this gene may be early driver events in the development of ESCC. In HNSCC, the CASP8 inactivating mutations have been reported to confer cells the ability of resistance to death receptor [50]. However, recent studies have reported that the function of CASP8 in cancer represents a complex double-edged sword, and its specific role in ESCC remains to be further elucidated [51, 52]. While most genome-scale rearrangements are thought to be neutral, some give rise to oncogenic gene fusions [32]. Gene fusions are potential targets for personalized therapeutics, and may also serve as potential prognostic and diagnostic biomarkers [53]. The current study presented the gene fusion landscape of esophageal squamous precursors and carcinoma. However, further studies are needed to determine whether these gene fusions are drivers or passengers.
Our study found that a high level of ploidy alterations can effectively distinguish between the two histologically different cohorts, suggesting that it may be an adaptive mechanism enabling cell survival under selective pressure. Aneuploidy is closely associated with increased genomic instability [54], thus aligning with the observation that ESCC is characterized by high levels of genomic instability [36]. Similarly, the transition from carcinoma in situ to LSCC is associated with a high level of chromosomal instability, which was identified as an early marker in this progression [55, 56]. Previous research comparing the genomic landscapes of early-stage and late-stage ovarian high-grade serous carcinoma (HGSC) also observed a significant increase in overall ploidy in late-stage tumors compared to their early-stage counterparts [57]. Moreover, genomic instability and aneuploidy may facilitate metastatic progression by inducing epithelial-to-mesenchymal transition (EMT), thereby promoting tumor invasion [58]. High levels of aneuploidy and genomic instability also correlate with the expression of immune evasion markers [59] and with poor prognosis [58, 60]. Our results suggest that a high level of ploidy confers cells with the ability to outcompete the original diploid dysplastic cells. This may result in phenotypic changes, such as the ability to infiltrate the basement membrane, which can be recognized as a distinct pathological change by histological examination.
Long-term follow up in our study revealed that patients with precancerous lesions characterized by elevated ploidy alterations displayed a greater risk of recurrence following ESD treatment. This result provides further evidence that intraepithelial neoplasia characterized by a high level of ploidy alterations has an increased risk for malignancy. Furthermore, the identification of individuals with high-risk dysplasia may facilitate more targeted and effective endoscopic surveillance and monitoring strategies. In summary, ploidy alterations appear to be closely associated with malignant transformation and with local recurrence of esophageal dysplasia.
Our comprehensive molecular profiling revealed distinct temporal phases in ESCC tumorigenesis. It also identified critical early-stage alterations that initiate malignant transformation, followed by subsequent transitional genomic events that are associated with tumor progression and phenotypic evolution. However, the findings of our study are subject to several limitations. First, due to the very small tissue volume of dysplasia samples, direct validations using these specimens were not feasible in the present study. And due to the limited samples number, we may not be able to capture a broader spectrum of genomic variations. We therefore plan to accrue a prospective clinical cohort of premalignant samples to replicate our findings. Second, although the current study includes long-term clinical follow-up validation and confirmation through genomic and transcriptomic analyses in independent cohorts, it is limited by the absence of molecular experiments. The functional effects of MUC5B mutations and CASP8 deletions still require mechanistic molecular experiments in further studies. Third, our next-generation sequencing analysis was restricted to the genomic and transcriptomic level, without the incorporation of epigenetic data. To overcome these constraints, subsequent investigations should employ multi-omics strategies that combine whole-genome, transcriptome, epigenome, and proteome analyses across a well-curated sample cohort.
In summary, this study comprehensively characterized the genomic alterations in ESCC and precancerous lesions. The cancer hallmark of genomic instability was prevalent even in the precancerous stages, and increased further during the transition from precursor to invasive cancer (Fig. 6). Mutations in TP53 and MUC5B, as well as the deletion of CASP8, may represent early driver events in carcinogenesis that precede emergence of the APOBEC mutagenesis pattern. In addition, ploidy alterations confer a selective advantage to genomically unstable cells, promoting their progression toward malignant transformation and local recurrence. These findings provide new insights into the genomic evolution of ESCC and highlight potential avenues for early detection and targeted intervention in esophageal carcinogenesis.
 Fig. 6.
Fig. 6.
Preliminary model of the evolution from dysplasia to ESCC. The schematic diagram of esophageal precancerous lesion and cancerous tissue in Fig. 6 was generated by ChatGpt (version 5.0; OpenAI, San Francisco, CA, USA).
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
HA performed the advanced bioinformatics analysis and wrote the manuscript. XC helped to verify the analysis results. GW participated in the experimental design and provided the dysplasia specimens used in this research. LX provided ESCC specimens for the research and conducted pathological diagnosis of all the samples. XZ performed basic bioinformatics analysis of data. JL assisted with literature retrieval and proofread the manuscript. SC took the lead in designing the study and overseeing the experimental framework. TX played a critical role in data analysis and drafting the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
This study was performed in accordance with the Declaration of Helsinki and was approved by the Institutional Review Board of Cancer Hospital, Chinese Academy of Medical Sciences (Ethic Approval Number: 12-71/605). A written consent was signed by the patients or their families/legal guardians.
We are grateful to Professor Kaitai Zhang and Lin Feng from the Cancer Hospital, Chinese Academy of Medical Sciences for their guidance on the analytical approach. We are grateful to Haonan Gu for his assistance in collecting the clinical follow-up data. We sincerely thank all the researchers and study participants for their contributions.
This work was supported by the National Natural Science Foundation of China (No. 82372718), the National Key R&D Program of China (No. 2023YFC3503205), and the Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences (No. 2023-I2M-2-004).
The authors declare no conflict of interest. Xiuli Zhu is an employee of Geneplus-Beijing Institute, the judgments in data interpretation and writing were not influenced by this relationship.
During the preparation of this work we used ChatGpt (version 5.0; OpenAI, San Francisco, CA, USA) to generate a schematic diagram of esophageal precancerous lesion and cancerous tissue in Fig. 6. After using this tool, we reviewed and edited the content as needed and takes full responsibility for the content of the publication.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBL41107.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.