
- Academic Editor
Epidemiological data show a strong connection between type 2 diabetes mellitus (T2DM) and metabolic-associated fatty liver disease (MAFLD). In recent years, the prevalence of both conditions has been rising simultaneously. When T2DM and MAFLD occur together, patients face a significantly higher risk of glucose and lipid metabolic disorders, with fatty liver more likely to progress to fibrosis or even malignancy. The underlying mechanisms are complex, involving multiple factors such as inflammatory responses, insulin resistance (IR), and cellular aging. Ferroptosis, a newly identified form of programmed cell death characterized by iron accumulation and lipid peroxidation, plays a crucial role in the development of T2DM and MAFLD, drawing significant attention. Current research suggests that ferroptosis contributes to the progression of these two diseases. However, the exact mechanisms of ferroptosis in T2DM-related MAFLD remain unclear. This review summarizes recent advances in ferroptosis research related to T2DM and MAFLD and highlights several potential therapeutic drugs and compounds targeting ferroptosis, aiming to provide a theoretical basis for their clinical application. Additionally, intracellular iron overload, elevated reactive oxygen species levels, and lipid peroxidation are closely associated with ferroptosis. Studies have shown that certain antidiabetic medications (e.g., metformin, pioglitazone, and liraglutide) may slow the progression of MAFLD by inhibiting ferroptosis. Furthermore, experimental studies targeting FerroTerminator1 (FOT1) have demonstrated promising therapeutic value for MAFLD and insulin resistance, suggesting that targeting ferroptosis could be an effective strategy for treating T2DM-related MAFLD.
Metabolic-associated fatty liver disease (MAFLD) is a disease with steatosis of liver cells as its basic pathological feature. With the progression of the disease, steatohepatitis, fibrosis and even liver cancer may occur, and it has become the most common chronic liver disease in the world [1]. Epidemiological investigation shows that the prevalence of MAFLD is increasing year by year and showing a trend of younger age, and the global prevalence of MAFLD has increased by about 50% in the past three decades [2]. Diabetes mellitus (DM) is a lifelong metabolic disease with multiple causes. Prolonged exposure to high glucose levels and metabolic disorders can lead to an increase in oxidative stress and reactive oxygen species (ROS) levels within the cardiac, hepatic, and other systemic tissues of patients. These factors may subsequently induce cell death, thereby causing organ tissue damage [3, 4]. Studies have found that compared with the general population, patients with type 2 diabetes mellitus (T2DM) have a more than 2-fold increased risk of developing MAFLD [5, 6]. According to epidemiological surveys, MAFLD exists in more than 25% of adults worldwide, and the prevalence of MAFLD in DM patients is as high as 50%–70% [1, 7]. From the perspective of the influence of DM on the systemic system, MAFLD belongs to the liver complication of DM, and a study has shown that T2DM is an important indicator to clinically predict the progression of MAFLD to MAFLD and cirrhosis [8]. In addition, prior research has highlighted that MAFLD is also a significant risk factor for T2DM. Patients with both T2DM and MAFLD may face greater challenges in managing their blood glucose levels, experience more severe lipid metabolism disturbances, and see an acceleration in the progression of diabetes-related organ damage. Furthermore, there exists a complex interplay between these two conditions [9, 10, 11]. These conditions are closely linked pathophysiologically, with insulin resistance (IR) and lipid metabolism disorders as the central mechanisms. Hepatic steatosis and inflammation worsen hepatic IR, impairing glucose regulation and promoting T2DM. T2DM’s hyperglycemic and hyperinsulinemic state stimulates hepatic lipogenesis and adipose tissue IR, increasing free fatty acid (FFA) flux to the liver. IR drives lipolysis in adipose tissue and hepatic lipid synthesis, leading to systemic lipid metabolism disorders. Lipotoxicity and its induced inflammation further perpetuate IR and organ dysfunction, creating a vicious cycle between the two diseases [12].
At present, the in-depth exploration of the pathogenesis of T2DM and MAFLD and the search for potential therapeutic targets have become a significant area of focus for global public health in recent years.
Ferroptosis is a newly discovered form of cell death in recent years.
Characterized by iron accumulation and lipid peroxidation during the process,
this type of cell death is iron-dependent, setting it apart from other modes like
apoptosis, autophagy, and pyroptosis [13]. Recent studies have highlighted the
connection between ferroptosis and diabetes [14]. Ferroptosis can impair the
function of pancreatic
Iron is crucial for maintaining normal human physiological functions, including hemoglobin synthesis for oxygen supply, cell metabolism, DNA repair and synthesis, and regulating immune function by promoting immune cell growth and differentiation [17, 18]. The body obtains iron from various sources, including heme iron, non-heme iron from food, and iron ions released from decomposed red blood cells [17].
Under normal conditions, iron ions are primarily used for hemoglobin synthesis, with excess stored as ferritin (FTN) and hemosiderin in the liver and macrophages. When iron demand increases, stored iron is released and distributed to tissues via the membrane transporter ferroportin (FPN) [19, 20]. At the systemic level, hepcidin, a protein secreted by hepatocytes, plays a key role. It binds to FPN on the surfaces of intestinal cells, macrophages, and hepatocytes, which leads to their internalization and degradation. This process helps inhibit iron absorption and release. When the body is in an iron-deficient state, hepcidin expression decreases, allowing for enhanced iron transport and absorption [21]. Intracellular iron homeostasis is maintained by the iron-regulatory protein (IRP)-iron response element (IRE) system, which regulates iron metabolism gene expression by binding to conserved mRNA motifs. When intracellular iron is low, the system promotes IRP1 synthesis and inhibits FPN expression, enhancing iron intake. Once cells are iron-saturated, transferrin receptor 1 (TFR1) expression decreases, limiting further iron uptake [18, 22].
Under normal conditions, iron ion storage and transport maintain a dynamic balance for human iron homeostasis [23]. Intracellular iron is stored in labile iron pools (LIP) and FTN [24]. When intracellular iron levels are excessive, this balance is disrupted, FTN degrades via autophagy-lysosomes mediated by nuclear receptor coactivator 4 (NCOA4), releasing Fe2+. Accumulated Fe2+ triggers Fenton reactions, producing toxic reactive oxygen species that damage proteins, DNA, and cell membranes, ultimately inducing ferroptosis [25]. Research shows that intracellular FTN levels influence ferroptosis sensitivity. Higher FTN levels reduce the labile iron pool, protecting cells from ferroptosis; conversely, lower FTN levels increase the labile iron pool, promoting ferroptosis [26, 27]. This occurs because FTN stores iron in an inert form, inhibiting its involvement in oxidative processes. Study also indicates Prominin-2 promotes FTN-containing exosome formation, facilitating iron export and inhibiting ferroptosis [28].
Iron metabolism imbalance serves as the primary trigger of ferroptosis, wherein free iron catalyzes lipid peroxide generation via the Fenton reaction, ultimately inducing cell death. Ferritin, the predominant intracellular iron storage protein, plays a pivotal role in regulating free iron levels and thereby modulating ferroptosis. Ferritin is a multi-subunit protein complex composed of heavy chains (FtH) and light chains (FtL), primarily functioning to sequester free iron and prevent its involvement in oxidative stress reactions. FtH exhibits ferrous oxidase activity, capable of oxidizing Fe2⁺ to Fe3⁺ and storing it within the protein shell, thereby reducing the labile iron pool (LIP) in cells [29]. This storage mechanism not only maintains iron homeostasis but also mitigates ROS production by inhibiting the Fenton reaction, thus protecting cells from oxidative damage [30].
Several studies have highlighted a potential link between ovarian cancer and ferritin. Under non-adherent culture conditions, HEY and PEO1 cells (both are ovarian cancer cell line) upregulate FtH1 expression to reduce LIP, thereby reducing ROS levels and avoiding ferroptosis. Notably, PEO1 cells exhibit a more pronounced increase in FtH1 expression (8.4-fold) and a more significant reduction in LIP (9-fold), indicating stronger resistance to ferroptosis. When FtH1 is knocked down, the spheroid formation ability of PEO1 cells is notably impaired, although this does not enhance sensitivity to ferroptosis, this suggests that compensatory antioxidant mechanisms (e.g., glutathione peroxidase 4 (GPX4) or nuclear factor erythroid 2-related factor 2 (NFE2L2) pathways) may be involved. By binding free iron and inhibiting lipid peroxidation, ferritin (particularly FtH) emerges as a critical regulatory node in ferroptosis [31]. However, abnormally elevated ferritin levels may indicate iron overload in tissues. Selective degradation of ferritin through ferritinophagy releases substantial amounts of iron ions, markedly increasing the labile iron pool, which represents a key step in triggering ferroptosis [32]. While ferritin degradation promotes ferroptosis, high ferritin expression may offer protective benefits in certain diseases. Further research is needed to clarify the molecular mechanisms governing dynamic ferritin regulation, paving the way for precise therapies targeting ferroptosis-related conditions.
Lipid peroxidation primarily causes oxidative damage to organelles and plasma membranes through spontaneous lipid oxidation and enzyme catalysis, driving ferroptosis. Polyunsaturated fatty acids (PUFAs) are key substrates in this process [33]. Enzyme-catalyzed lipid peroxidation requires long-chain acetyl-CoA synthetase 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3). These enzymes catalyze the esterification of free PUFAs into phospholipids, leading to their accumulation in cell membranes. Subsequently, lipoxygenase catalyzes membrane disruption, triggering ferroptosis [34].
Current studies show that ACSL4-mediated long-chain fatty acid metabolism is crucial for initiating ferroptosis [35]. Fe2+ participates in the Fenton reaction, leading to lipid oxidation and the production of hydroxyl free radicals, which oxidize PUFA-containing phospholipids, causing peroxides and ferroptosis [36]. Research on fibrosarcoma cells treated with four ferroptosis inducers revealed that lipid peroxidation initially accumulates in the endoplasmic reticulum (ER), followed by membrane peroxidation and morphological changes. This indicates the ER is a key target for lipid peroxidation, guiding the development of ferroptosis inhibitors and inducers [37].
Under normal conditions, intracellular lipid peroxides are cleared primarily by the glutathione peroxidase 4/glutathione (GPX4/GSH) axis. GPX4 reduces lipid peroxides to phospholipids using GSH [38]. GSH synthesis depends on System Xc–, which exchanges extracellular cystine for intracellular glutamate [39, 40], Cystine is then reduced to cysteine and incorporated into GSH. Depletion of GSH and inhibition of GPX4 lead to lipid peroxide accumulation, ROS generation, and ferroptosis. The System Xc–/GSH/GPX4 pathway is a key ferroptosis mechanism [41], Beclin 1 binds to System Xc– and inhibits its activity, reducing GSH levels and promoting oxidative stress and ferroptosis [42]. P53 suppresses System Xc– activity by downregulating SLC7A11, leading to ferroptosis [43]. In addition, researchers identified an antioxidant pathway involving ferroptosis suppressor protein 1 (FSP1) and coenzyme Q10 (CoQ10). CoQ10 inhibits lipid peroxide formation, while FSP1 reduces CoQ10 to eliminate lipid peroxidation and inhibit ferroptosis [44, 45]. It was found that ginsenoside Rg1 activates the FSP1/CoQ10 axis, thereby inhibiting lipopolysaccharide-induced lipid peroxidation in human kidney cell line (HK-2) cells and prevents ferroptosis in renal tubular cells [46, 47]. Dihydroorotate dehydrogenase (DHODH), a CoQ10-reducing flavin protein similar to FSP1, inhibits ferroptosis in the mitochondrial membrane and regulates cellular sensitivity to the GPX4 inhibitor RAS selective lethal 3 (RSL3) [48, 49].
IR plays a crucial role in the development of T2DM and MAFLD. Individuals with T2DM often experience peripheral IR, which can result in disorders of glucose and lipid metabolism, this leads to increased fat breakdown, higher levels of FFA, and eventually hepatic steatosis [50]. Current studies show that IR is positively correlated with MAFLD development, liver fibrosis, and can promote hepatocyte steatosis, progressing to MAFLD and liver fibrosis [51]. An increasing number of studies suggest that IR is closely linked to iron overload, and disruptions in iron balance may contribute to the development of IR [52, 53] (Fig. 1). High-iron diets may alter insulin signaling in the liver and muscles, resulting in elevated hepatic glucose production in mice with excessive iron levels [53]. Research shows that treating iron overload can help improve IR. A study on animals has found that using iron chelators can make obese models more sensitive to insulin [54].
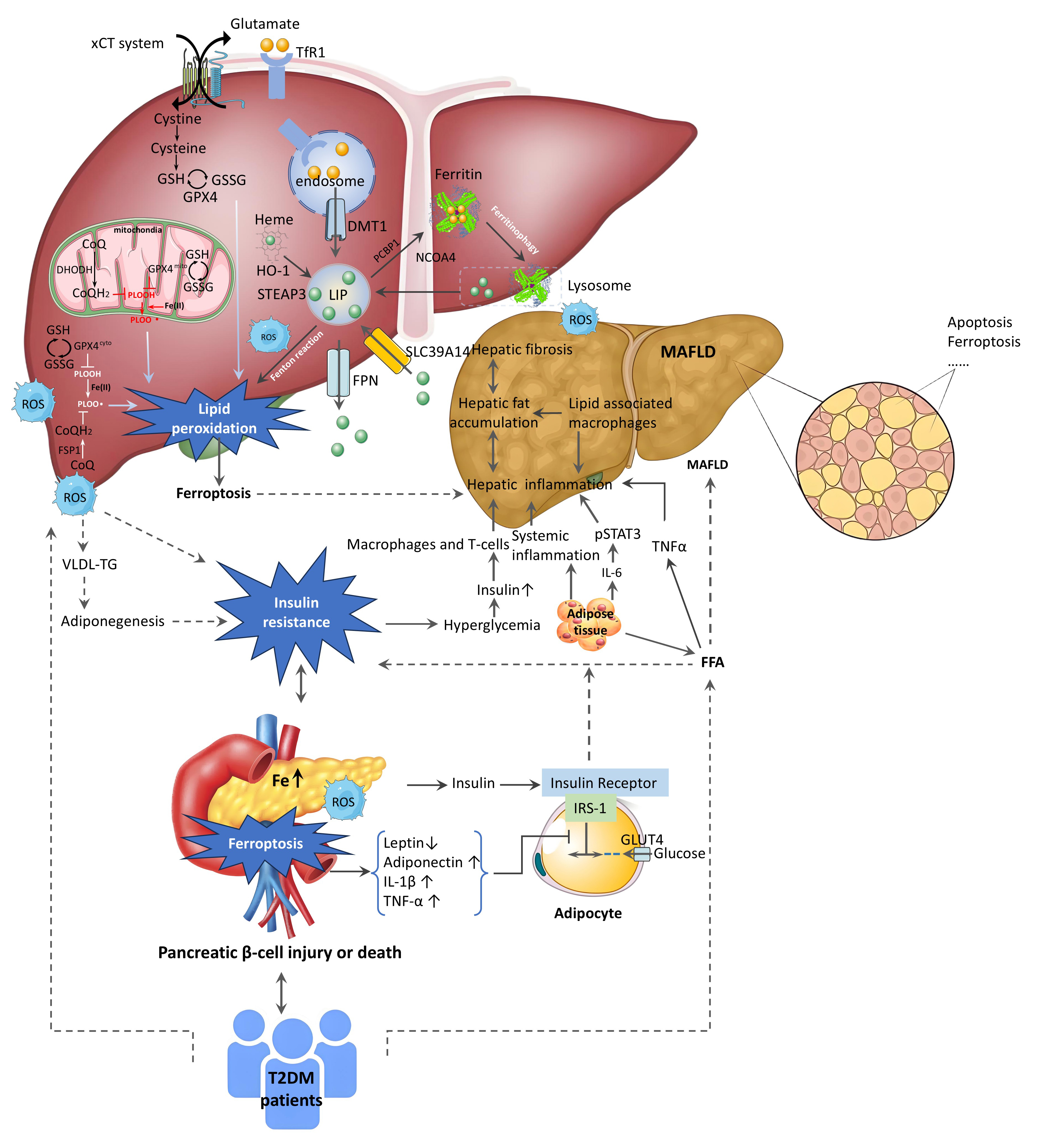 Fig. 1.
Fig. 1.
The regulatory mechanisms of ferroptosis in MAFLD and the
relationship between T2DM and the progression of MAFLD. The connection between
MAFLD and T2DM is mediated through IR, ROS, and LPO. MAFLD leads to liver
steatosis, lipogenic changes, declining function, and inflammation, worsening IR
and driving T2DM progression. Elevated FFA levels in MAFLD exacerbate liver IR
and fat accumulation. In T2DM, IR increases insulin secretion, intensifying FFA
synthesis. Higher hepatic fatty acids cause hepatocyte dysfunction, inflammation,
oxidative stress, and accelerate MAFLD development. Inflammation induces
oxidative stress, leading to cell death, tissue damage, mitochondrial
dysfunction, and excessive FFA accumulation, impairing insulin signaling and
sensitivity. The liver regulates glycolipid metabolism and iron storage; recent
studies link these functions to ferroptosis. Iron overload damages mitochondria,
increases ROS, reduces adiponectin receptor expression, and worsens IR,
collectively driving disease progression.
Iron overload increases intracellular ROS production, which impairs glucose
uptake and insulin sensitivity in muscle and fat [55]. Elevated ROS levels cause
oxidative stress, contributing to IR pathogenesis, particularly through
mitochondrial damage [56]. FFA are also key factors in IR. Palmitic acid (PA)
upregulates TFR1, promoting endoplasmic reticulum stress and calcium depletion,
leading to iron overload, mitochondrial damage, and increased ROS production.
This ultimately reduces insulin sensitivity and causes IR. After knocking out the
TFR1 gene, PA failed to induce iron overload and IR in cells, and iron chelating
agents significantly reduced PA-induced IR [37, 57]. Research shows that
increasing mitochondrial NEET protein (mitoNEET) expression regulates iron levels
in heart and skeletal muscle cells, reducing harmful ROS production and
protecting against iron overload damage [58], iron overload can lead to
ferroptosis and IR by inhibiting the janus kinase 2/signal transducer and
activator of transcription 3/solute carrier family 7 member 11
(JAK2/STAT3/SLC7A11) pathway. However, the iron chelator deferasirox (DFX) helps
reduce glycogen and GSH levels in liver tissue and HepG2 cells, thereby
alleviating iron overload-induced IR [59]. Autophagy disorders contribute to IR
development. Iron overload inhibits the mechanistic target of rapamycin complex
1–UV radiation resistance associated gene (mTORC1-UVRAG) pathway, leading to
autophagy defects and promoting IR. Salubrinal, an eukaryotic translation
initiation factor 2 subunit alpha (eIF2
At present, a number of studies have shown that iron overload is closely related to adiponectin. Adiponectin is a protein secreted by adipocytes that can enhance insulin sensitivity and regulate glycolipid metabolism. There is a negative correlation between adiponectin levels and IR. The decreased expression level or signal transduction of this protein can cause impaired insulin signal transduction and glucose tolerance, thereby promoting the occurrence and aggravation of IR [62, 63]. Adiponectin requires binding to cell surface receptors to mediate its effects. Increased ROS in iron-overloaded muscle cells causes forkhead box protein O1 (FOXO1) phosphorylation due to oxidative stress, inhibiting adiponectin receptor transcription and reducing receptor expression, leading to adiponectin resistance [64, 65]. A Study shows adiponectin restores carnitine palmitoyltransferase 1 (CPT1) activity in gestational diabetes mice, correcting ferroptosis and improving placental damage [66]. Previous research indicates osteocalcin increases adiponectin expression in adipocytes, enhancing insulin sensitivity. Spanish researchers found iron overload decreases circulating osteocalcin levels in cell experiments. Cross-sectional studies confirm a negative correlation between ferritin and osteocalcin/adiponectin levels, suggesting osteocalcin-mediated adiponectin effects on IR may represent a new mechanism [67].
In summary, iron overload contributes to IR via multiple mechanisms, including ROS generation, mitochondrial dysfunction, and inhibition of adiponectin signaling. Conversely, IR may further exacerbate iron overload, thereby establishing a vicious cycle. This interaction represents a critical link between T2DM and MAFLD. Notably, the majority of existing evidence originates from animal models or in vitro experiments. Clinical studies that directly demonstrate iron overload as the primary driver of IR and MAFLD progression in T2DM patients, while controlling for confounding factors such as obesity and inflammation, remain scarce. The identification of the osteocalcin-adiponectin-IR axis as a potential novel mechanism is intriguing; however, further in-depth functional investigations and clinical validation are warranted.
Ferritin, an essential protein for iron storage, is crucial in diabetes
pathophysiology. Study shows higher serum ferritin levels in uncontrolled T2DM
individuals, correlating positively with glycated hemoglobin and fasting blood
glucose. This suggests iron overload may indicate metabolic disorders in
hyperglycemic conditions [68]. Chronic inflammation in diabetes triggers
hepcidin production, reducing iron absorption and release, and causing iron
accumulation in the liver and pancreas. Ferritin FtH1 expression protects
The liver serves as a pivotal organ for glucose and lipid metabolism, as well as iron homeostasis. Serum ferritin acts as the primary protein responsible for iron storage in the liver, and elevated ferritin levels are frequently observed in MAFLD. As a chronic inflammatory condition, MAFLD induces fat-laden hepatocytes and infiltrating immune cells to secrete substantial amounts of pro-inflammatory cytokines. These cytokines subsequently stimulate hepatocytes and macrophages to synthesize and release ferritin, thereby contributing to elevated serum ferritin levels. A cross-sectional study involving 523 patients with T2DM revealed that individuals with hyperferritinemia exhibited significantly higher prevalence rates of MAFLD and nonalcoholic steatohepatitis (NASH) compared to those without hyperferritinemia. After adjusting for age, gender, obesity, and insulin usage, hyperferritinemia persisted as an independent predictor of MAFLD and NASH [70]. Investigations into the relationship between elevated SF levels and T2DM/MAFLD highlight clinical significance; however, the interpretation of SF as an inflammatory marker warrants caution. It remains unclear whether the elevation in SF represents the cause or consequence of iron overload or merely an accompanying phenomenon associated with inflammation.
Owing to insulin insufficiency or resistance, T2DM triggers the mobilization and
decomposition of adipose tissue, leading to lipid metabolic disorders and
increased FFA production, which are subsequently metabolized and decomposed by
the liver [71]. The accumulation of lipids in liver tissues and increased FFA
uptake by hepatocytes boost mitochondrial
The liver is a remarkable organ where glycogen synthesis, decomposition, and gluconeogenesis take place, playing a crucial role in maintaining stable glucose levels. As an important iron storage site, the liver synthesizes and secretes hepcidin, which helps regulate circulating iron levels and contributes to the balance of iron metabolism in human bodies [78]. We currently understand that there is a complex relationship between iron overload, T2DM, and MAFLD, where liver cells play a crucial role. In fact, the liver has a high iron content, and iron overload can cause significant damage to it. Iron overload may accelerate MAFLD progression to liver fibrosis and cirrhosis, while, MAFLD can worsen iron overload [79]. Liang and colleagues examined liver iron content in 494 MAFLD patients, finding positive correlation between liver iron accumulation and disease progression. By integrating findings from various mouse models of MAFLD disease, researchers concluded that liver iron accumulation triggers ferroptosis via the c-Myc-ACSL4 pathway, accelerating disease progression [61]. Liver macrophages store iron and drive inflammation in MAFLD. Zhang et al. [80] found that neutrophil cytoplasmic factor expression in macrophages induces hepatocyte hepcidin production via phospholipid peroxides, causing macrophage ferroptosis. The loss of iron-laden macrophages leads to an influx of inflammatory cells, worsening liver inflammation and accelerating MAFLD progression [80] (Fig. 1).
Metabolic abnormal iron overload syndrome is characterized by iron overload and MAFLD, both linked to metabolic syndrome and commonly seen in obese individuals and those with T2DM [81]. The iron overload is often caused by an unhealthy diet and worsened by genetic and environmental factors. While a direct causal relationship with metabolic syndrome remains unclear [82], T2DM increases oxidative stress and ROS levels in the liver, causing cell ferroptosis and liver damage, which promotes fatty liver development [83]. At night, gluconeogenesis is more active, and iron regulates the circadian rhythm of liver glucose production. Iron overload inhibits glycogen synthesis, weakens insulin’s suppression of gluconeogenesis, and leads to disordered glucose metabolism and elevated glucose levels [84, 85]. The finding suggests that iron overload disrupts the circadian rhythm of liver glucose metabolism holds promising clinical significance. However, translating this insight into practical intervention strategies for managing blood glucose fluctuations in T2DM patients remains an area to be further explored and developed.
Iron overload significantly contributes to IR, a common risk factor closely associated with T2DM and MAFLD. Further studies show that IR not only worsens iron overload but also creates a vicious cycle [86]. Similarly, MAFLD and IR interact in a comparable manner, IR causes adipose tissue to release excess FFA into the liver, promoting lipid accumulation and fatty liver formation. In turn, fatty liver impairs insulin signaling via mechanisms like pro-inflammatory factor secretion and excessive ROS production, exacerbating IR [87, 88].
Over the past few decades, understanding of MAFLD pathogenesis has raised hopes
for effective treatment. To date, only Resmetirom, a selective hepatic thyroxine
receptor
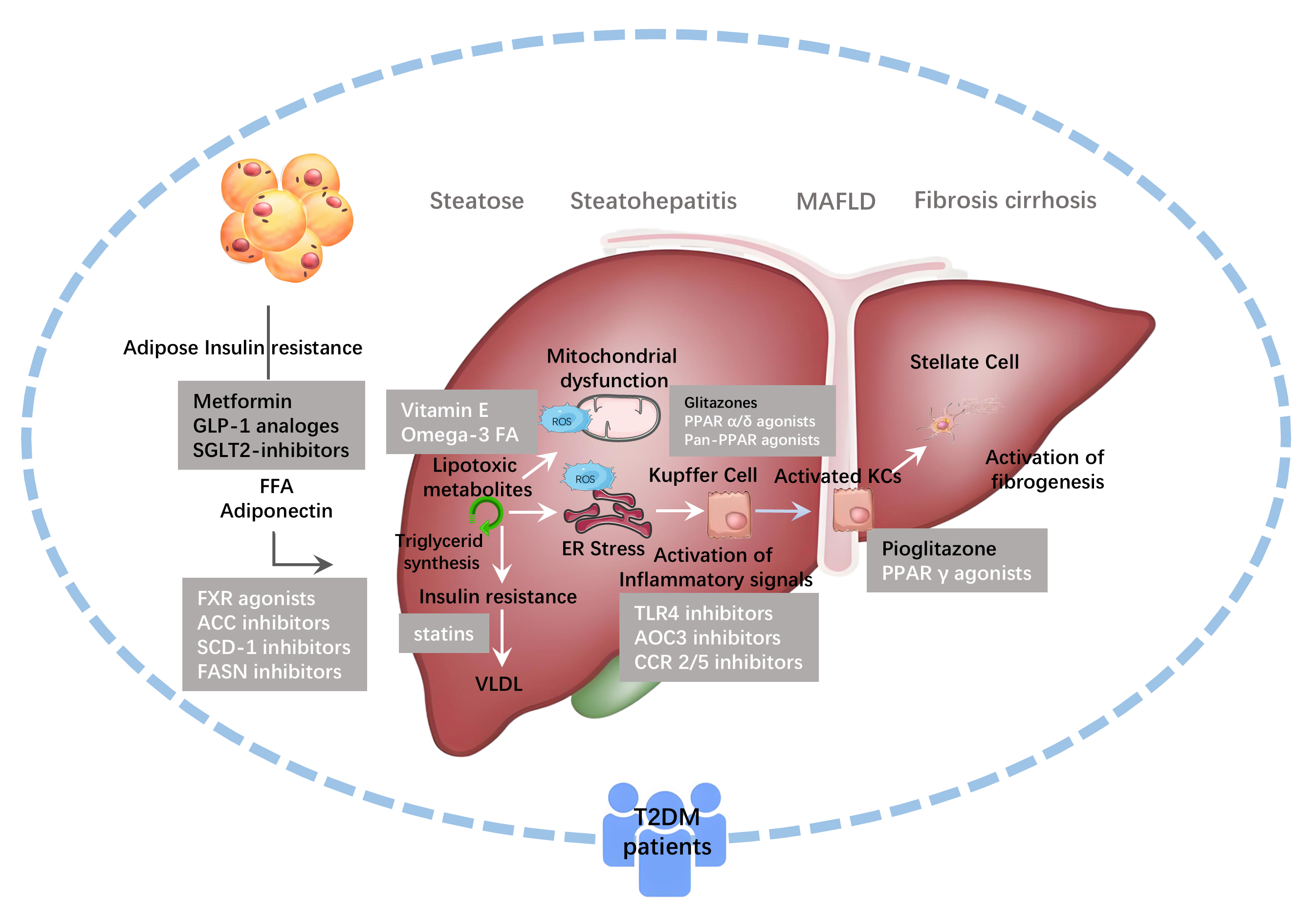 Fig. 2.
Fig. 2.
Drugs Targeting Ferroptosis in MAFLD. The drugs currently used
in the clinical treatment of MAFLD/NASH include: nuclear receptor agonists such
as farnesoid X receptor (FXR) agonists, peroxisome
proliferator-activated receptor (PPAR) agonists, chemokine receptor inhibitors,
thyroid hormone receptor-
| Drugs | Types | Targets | Mechanism |
| Metformin | Biguanides | xCT, GPX4 | Upregulate xCT and GPX4, decrease ACSL4, inhibit ferroptosis |
| Semaglutide | GLP-1RA | Nrf2 | Active Nrf2, inhibit lipid peroxidation |
| Liraglutide | GLP-1RA | Nrf2, AMPK | Active Nrf2 and AMPK, inhibit LPO and ferroptosis |
| Dulaglutide | GLP-1RA | Nrf2 | Upregulate KLB levels |
| Pioglitazone | Thiazolidinediones | ACSL4 | Inhibit ACSL4, inhibit LPO and ferroptosis |
| Pioglitazone | Thiazolidinediones | mitoNEET | Stabilize mitoNEET, reduce LPO and ferroptosis in hepatocytes |
| Deferoxamine | Iron chelator | Free iron | Chelate iron, inhibit Fenton reaction |
| Deferiprone | Iron chelator | Free iron | Chelate iron, inhibit Fenton reaction |
| Deferasirox | Iron chelator | Free iron | Chelate iron, inhibit iron overload, inhibit ferroptosis |
| Ferrostatin-1 | Antioxidant | PUFA, ROS | Scavenge ROS, inhibit LPO |
| Trolox | Antioxidant | PUFA | Inhibit Lipid peroxidation and ferroptosis |
| Water-soluble Vitamin E | |||
| Derivatives | |||
| Liproxstatin-1 | Ferroptosis inhibitor | ROS, Nrf2, GPX4 | Scavenge ROS, upregulate GPX4, active Nrf2, inhibit LPO |
*ACSL4, acyl-CoA synthetase long-chain family member 4; AMPK, Adenosine 5′-monophosphate (AMP)-activated protein kinase; xCT, antiporter of cystine and glutamate, system Xc–; GLP-1RA, glucagon-like peptide-1 receptor agonist; GPX4, glutathione peroxidase 4; LPO, lipid peroxidation; mitoNEET, CDGSH Iron-Sulfur Domain 1; Nrf2, nuclear factor erythroidderived 2-like 2; ROS, reactive oxygen species; PUFA, polyunsaturated fatty acid; MAFLD, Metabolic-associated fatty liver disease; mitoNEET, mitochondrial NEET protein; KLB, klotho beta.
Metformin (MET) serves as the first-line therapy for T2DM, and clinical studies have confirmed its ability to enhance the therapeutic efficacy in MAFLD (Fig. 2). Its effectiveness is linked to its anti-inflammatory, antioxidant properties, and its role in regulating autophagy and necroptosis [89, 90]. Recent research shows that metformin slows the progression of MAFLD by mediating ferroptosis in hepatocytes. It was found that two ferroptosis inducers triggered ferroptosis in alpha mouse liver 12 (AML12) cells, while metformin improved cell viability and survival rates after intervention [91]. Metformin alleviated palmitic acid (PA)-induced ferroptosis in AML12 cells and T2DM mouse hepatocytes by reducing intracellular iron overload and ROS levels. This study also highlighted that metformin increased antiporter of cystine and glutamate (xCT) and GPX4 levels in hepatocytes, decreased ACSL4 expression, thereby improving MAFLD through regulation of ferroptosis via the xCT/GPX4/ACSL4 axis [91]. Similarly, Studies have found that metformin activates the AMPK pathway, reducing lysosomal degradation of FPN. This increases FPN expression and alleviates iron overload and ferroptosis in liver cells. Metformin also significantly reduced liver weight and transaminase levels in high-fat diet-induced MAFLD mice, improving steatosis, inflammation, and fibrosis, thus preventing MAFLD progression [91, 92]. Unfortunately, this study observed that MET increases GPX4 expression in hepatocytes, but the precise mechanism of MET’s influence on the ferroptosis pathway remains unclear (Table 1). As a commonly prescribed first-line medication for managing T2DM in clinical practice, metformin is supported by extensive clinical evidence demonstrating its effectiveness in improving MAFLD. The mechanism related to ferroptosis might help explain, at least in part, its protective effects on the liver.
Glucagon-like peptide-1 (GLP-1) is a polypeptide hormone that stimulates insulin
secretion and inhibits glucagon release. Its physiological effects are mediated
through both receptor-dependent and non-receptor-dependent pathways. The latter
involves the mitochondrial function of small molecular fragments of GLP-1.
Extensive research has confirmed that various GLP-1 receptor agonists improve
fatty liver conditions and are closely associated with ferroptosis [93, 94] (Fig. 2). Specifically, studies on semaglutide have demonstrated its ability to inhibit
cellular ferroptosis by regulating klotho beta (KLB) expression and
subsequently modulating the adenosine monophosphate-activated protein
kinase/acetyl-CoA carboxylase/sirtuin 1/nuclear factor erythroid 2 related factor
2 (AMPK/ACC/SIRT1/Nrf2) signaling pathway (Table 1). Similarly, liraglutide and
dulaglutide have been shown to significantly upregulate KLB levels [95]. Recent
investigations indicate that liraglutide mitigates ferroptosis in hepatocytes via
the glycogen synthase kinase 3 beta/nuclear factor erythroid 2-related factor
2 (GSK3
Thiazolidinediones (TZDs) activate the peroxisome proliferator-activated
receptor (PPAR) signaling pathway in adipose tissue, reducing FFA production and
hepatic fat accumulation [99] (Table 1). Clinical studies show beneficial effects
on liver tissue. A cohort study of 207,367 patients found that TZD therapy helps
diabetic patients at risk for MAFLD [100]. Pioglitazone is effective in treating
MAFLD, especially in T2DM patients. Randomized controlled trials confirm that
pioglitazone reduces hepatocellular steatosis, inflammation, and fibrosis
progression [101, 102]. Research also indicates that TZDs, including pioglitazone,
inhibit ACSL4 expression, thus preventing lipid peroxidation and ferroptosis
induced by ferroptosis inducers [103] (Table 1). Moreover, pioglitazone
stabilizes mitoNEET expression and mitigates the inhibitory effect of ferroptosis
inducers on mitoNEET, reducing lipid peroxidation and ferroptosis in hepatocytes
[104] (Table 1). Pioglitazone is associated with risks like weight gain and fluid
retention, and it is not approved for MAFLD treatment in non-T2DM patients (Fig. 2). This somewhat limits its broad applicability as an anti-ferroptosis drug for
treating MAFLD. Investigating safer and more selective PPAR
Iron chelating agents play an essential role in clinical settings by helping manage iron overload. These agents work by forming stable complexes with iron ions, which promotes the body’s natural ability to excrete excess iron and reduces the overall iron burden. This process helps protect tissues and organs from damage caused by iron overload, such as lipid peroxidation and the production of harmful ROS. Iron chelators are particularly beneficial for patients who require long-term blood transfusions, offering vital support in managing iron levels. Among the currently approved options for clinical use, deferoxamine (DFO), deferiprone (DFP), and deferasirox (DFX) stand out as effective treatments [105] (Table 1). Emerging animal studies have revealed that iron chelating agents may help slow the progression of MAFLD and lessen the severity of non-alcoholic steatohepatitis (NASH). As a result, these agents hold considerable promise for treating MAFLD (Fig. 2, Table 1). In vitro experiments have shown that DFO significantly reduces iron-induced cell death triggered by PA and alleviates the detrimental effects of iron inducers on hepatocytes. In a NASH mouse model induced by methionine and choline-deficient diet, DFO intervention markedly decreases hepatic iron load, thereby effectively mitigating MAFLD severity. This improvement is evident in several key indicators, such as aminotransferase levels, lipid peroxidation, and GSH levels [76].
Deferrone has shown a remarkable role in the treatment of MAFLD (Fig. 2). In a mouse model of MAFLD induced by a high-fat and high-fructose diet, two weeks of deferrone intervention led to a marked reduction in hepatic iron levels and liver weight. Importantly, deferrone effectively inhibited the elevation of mitochondrial ROS levels. These experimental findings indicate that deferrone can significantly alleviate steatohepatitis symptoms in mice, providing a promising therapeutic option for MAFLD [106] (Table 1).
FerroTerminator1 (FOT1), a novel iron chelator with potent iron removal capabilities, not only effectively inhibits hepatic iron accumulation but also blocks ferroptosis induced by aberrant activation of the c-Myc-ACSL4 signaling pathway. Notably, FOT1 exhibited favorable safety profiles across various toxicity evaluation experiments, laying the groundwork for its future clinical application. Studies have shown that in multiple MAFLD mouse models, long-term FOT1 treatment significantly reduced liver iron levels, improved fibrosis scores, alleviated liver damage, and mitigated systemic IR compared to traditional iron chelators DFO and DFX (Fig. 2, Table 1). Additionally, FOT1 effectively suppressed the expression of genes associated with inflammation and fibrosis, demonstrating that prolonged FOT1 treatment can prevent and mitigate the progression of MAFLD in mice [61] (Table 1). However, the efficacy of iron chelation therapy in preventing and treating MAFLD remains limited to animal models. The long-term use of iron chelators carries a notable risk of causing iron deficiency, which could potentially lead to anemia. As such, it would be valuable to conduct further clinical and translational research to confirm their therapeutic benefits while ensuring patient safety (Fig. 2).
As a class of powerful and highly selective inhibitors of ferroptosis, antioxidants can gently protect cells from iron-related lipid peroxidation by reducing the production of ROS and lipid peroxidation products [107, 108]. Similar to iron chelating agents, antioxidants have demonstrated significant therapeutic benefits for MAFLD in both animal and cell studies. When the body enters the senescence stage, liver cells face the dual challenge of an increased risk of ferroptosis and heightened lipid toxicity. In a study using a MAFLD senescence mouse model induced by coronary artery disease-high fat (CAD-HFD) diet, it was observed that after intervention with Ferrostatin-1 (Fer1), following the establishment of the MAFLD aging mouse model using a CAD-HFD diet, the expression of ACSL4 in liver cells was significantly reduced, along with a notable decrease in lipid peroxidation levels (Table 1). Importantly, Fer-1 effectively mitigated the adverse effects of aging on these critical parameters and markedly alleviated hepatic steatosis in aged mice. These positive changes made the livers of aged mice more similar to those of young and healthy mice, indicating that Fer-1 prevents MAFLD progression, a disease linked to aging [109] (Fig. 2, Table 1). In a mouse model of MAFLD induced by a high-fat and high-fructose diet, Liproxstatin-1 (Lpt-1) inhibited ferroptosis, reduced hepatic triglycerides and cholesterol, suppressed lipid synthesis and oxidation genes, alleviated IR, decreased mitochondrial ROS, and mitigated liver fibrosis. These findings indicate Lpt-1 effectively ameliorates steatosis and inhibits hepatitis progression in MAFLD mice [110].
Additionally, experimental evidence indicates that Trolox can inhibit ferroptosis in hepatocytes, reduce immune cell infiltration, and lower pro-inflammatory factors expression, improving MAFLD progression in mouse models [111]. Both animal and cellular studies indicate that iron chelators and antioxidants benefit MAFLD treatment, suggesting that inhibiting ferroptosis could be a promising new target (Table 1). However, practical application will require further drug development and clinical trials.
In summary, ferroptosis plays a pivotal role in the pathogenesis of T2DM-associated MAFLD, encompassing intricate interactions among iron overload, IR, elevated ferritin levels, lipid metabolic disorders, and hepatocyte injury. Regarding treatment strategies, commonly prescribed antidiabetic drugs have demonstrated the ability to inhibit ferroptosis, offering novel insights into their hepatoprotective mechanisms. Nevertheless, the critical evidence supporting the central role of ferroptosis primarily stems from cellular and animal studies, and its precise contribution in human T2DM with MAFLD requires confirmation through large-scale population-based studies and clinical trials.
In terms of therapeutic approaches, MET and GLP-1 receptor agonists, due to their well-established clinical profiles and robust efficacy data, currently represent the most promising intervention methods with practical translational potential. Although iron chelators and antioxidants have exhibited substantial effects in animal models, their clinical translation encounters significant challenges: the efficacy and safety of traditional iron chelators for treating non-anemic iron overload in MAFLD remain unclear, with the risk of inducing anemia; FOT1 and other emerging compounds lack human data, and their drugability requires further validation [61]. SF levels serve as crucial biomarkers of ferroptosis and can function as a comprehensive indicator to evaluate metabolic disturbances, inflammatory status, and the severity of liver injury in patients with T2DM and MAFLD. However, these levels should be interpreted in conjunction with inflammatory markers to differentiate between iron overload and inflammatory conditions.
Future research efforts should focus on further validating the role of ferroptosis in human diseases, discovering additional ferroptosis-related biomarkers and clinical testing methods, and exploring innovative, safe, and targeted therapeutic approaches based on the ferroptosis mechanism. By addressing these translational challenges, we can pave the way for targeted ferroptosis inhibition to truly become an effective clinical strategy for managing T2DM-associated MAFLD.
YS, FY, GW, FN designed the research study. GW, FY, YS, FN wrote the manuscript. YS, GW prepared the figures. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Thanks to all the peer reviewers for their opinions and suggestions.
This research were funded by National Administration of Traditional Chinese Medicine Fang Shuilin National Famous Veteran Traditional Chinese Medicine Expert Inheritance Studio, grant Number [2016] (42) issued by the Ministry of Education and Human Resources; Zhejiang Province Traditional Chinese Medicine Science and Technology Plan Project, grant number 2022ZB345; Key Research Projects of the Affiliated Hospital of Zhejiang Chinese Medical University, grant number 2022FSYYZZ22; Science and Technology Innovation Special Project of Jiaxing Science and Technology Bureau, grant number 2021AY30002 and 2024AY30006; Medicine and Health Science and Technology Plan Projects of Zhejiang Province, grant number 2023KY1227; Jiaxing Key Laboratory of Diabetic Angiopathy Research.
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.