


 , Sara Sossai 2, Jacopo Angelini 3, Gabriele Stocco 1,4,*
, Sara Sossai 2, Jacopo Angelini 3, Gabriele Stocco 1,4,* , Marianna Lucafò 5
, Marianna Lucafò 51 Department of Translational and Advanced Diagnostics, Institute for Maternal and Child Health I.R.C.C.S. Burlo Garofolo, 34137 Trieste, Italy
2 UOC Farmacia, Aulss 2 Marca Trevigiana, 31100 Treviso, Italy
3 Azienda Sanitaria Universitaria Friuli Centrale (ASU FC), 33100 Udine, Italy
4 Department of Medical, Surgical and Health Sciences, University of Trieste, 34129 Trieste, Italy
5 Department of Life Sciences, University of Trieste, 34127 Trieste, Italy
Abstract
The nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3 (NLRP3) inflammasome is a multiprotein complex fundamental for the secretion of pro-inflammatory cytokines during the innate immune response. NLRP3 dysregulation is implicated in the pathogenesis of several diseases, such as inflammatory bowel disease, arthritis, cancer, Alzheimer’s disease, and type 2 diabetes. The pharmacological modulation of NLRP3 by several compounds, which are fully described in this review, represents an important strategy to regulate inflammatory processes. Moreover, NLRP3 is also involved in drug-related adverse reactions, and its pharmacological modulation represents a rapid strategy to mitigate such adverse effects, as reported in this study. NLRP3 inflammasome activation is tightly regulated by post-transcriptional modifications and epigenetic factors, such as long non-coding RNAs (lncRNAs) and DNA methylation, as well as other interacting regulators. Recently, different studies have revealed the importance of NLRP3 levels in predicting drug response. In particular, the methylation of the NLRP3 promoter, which is associated with the inflammasome expression level, emerged as a new promising pharmacoepigenetic biomarker for the glucocorticoid therapy response in several inflammatory disease conditions.
Keywords
- NLRP3
- inflammasome
- personalization therapy
- biomarker
The human immune system comprises both adaptive immunity and innate immunity, with the latter serving as the first line of defense [1]. Nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3 (NLRP3) inflammasomes are intracellular multimeric protein complexes that activate pro-caspase-1, which is crucial for the initiation and control of inflammation. This activation is fundamental for the innate immune system activation and consequent inflammatory modulation. Notably, inflammasome dysregulation has been found to be involved in different inflammatory chronic conditions, including rheumatoid arthritis, systemic lupus erythematosus, type 2 diabetes, atherosclerosis, ischemic heart disease, liver diseases, amyotrophic lateral sclerosis, inflammatory bowel disease (IBD), Parkinson’s and Alzheimer’s disease [2]. Given that NLRP3 activity is tightly regulated, different pharmacological approaches have been developed to identify molecules capable of modulating its activity, thereby leading to inflammatory regulation.
The NLRP3 inflammasome formation, activation and modulation are straightly
related to the immune response activation and maintenance. In particular, the
innate immune response recognizes different pathogen-associated molecular
patterns (PAMPs, e.g., bacterial products like lipopolysaccharides) and derived
danger-associated molecular patterns (host DAMPs, e.g., uric acid crystals,
adenosine triphosphate (ATP)) through the pattern recognition receptors (PRRs) to
activate a physiological response against pathogens [3]. These PRRs subsequently
oligomerize to form inflammasomes [4], whose caspase-1-mediated activation
stimulates the secretion of proinflammatory cytokines, such as interleukin
(IL)-1
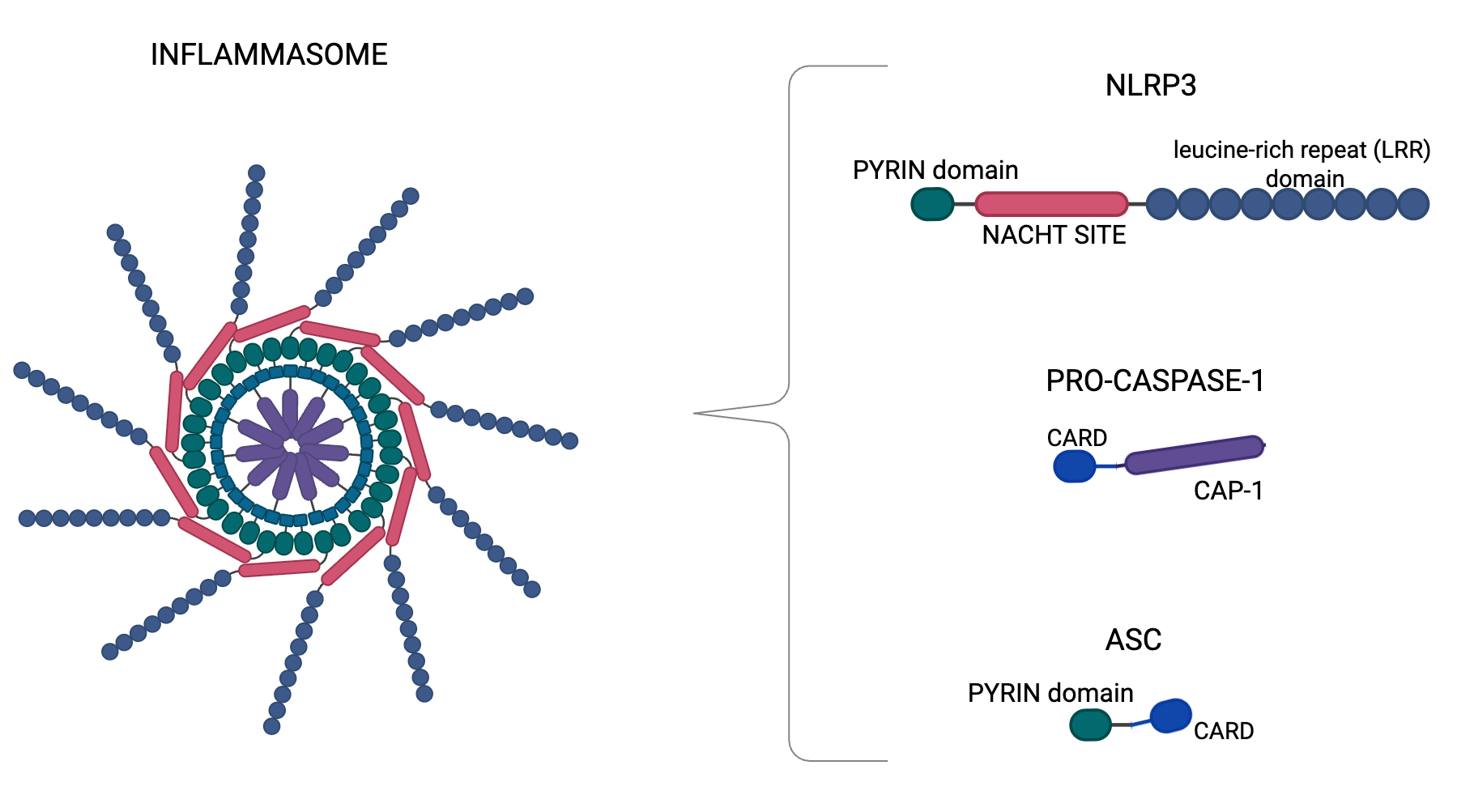 Fig. 1.
Fig. 1.
Inflammasome structure. Nucleotide-binding domain, leucine-rich–containing family, pyrin domain–containing-3 (NLRP3) inflammasome structure, which is a complex composed by NLRP3, apoptosis-associated speck-like protein (ASC), and procaspase-1. NLRP3 shows three regions: the pyrin domain in the amino terminus, the NACHT site, and the LRR (leucine-rich repeat) domain in the carboxy terminus. NLRP3 recruits ASCs through pyrin-pyrin domain interactions. In turn, procaspase-1 is recruited by ASC through Caspase Recruitment Domain (CARD)-CARD interactions to form the NLRP3-ASC-procaspase-1 (CAP-1) inflammasome. In particular, we generated Figs. 1,2 using BioRender.com (BioRender Inc., Toronto, Canada).
Since NLRP3 is the best characterized and studied isoform, this review focuses
on NLRP3 description. As previously specified for all inflammasome classes, NLRP3
can be activated by several stimuli, including PAMPs and DAMPs to form an
inflammasome that activates caspase-1 and promotes the maturation and secretion
of IL-1
NLRP3 activation is finely regulated, depends on different factors, and
comprises several steps. This complex mechanism is divided into two main phases:
the priming step and the activation step [10, 11]. The priming step consists of
increased cellular expression levels of pro-IL-1
In cells, NLRP3 activation can be induced by different stimuli, such as the
K+ and Cl– efflux due to PAMPs/DAMPs [12, 13], Ca2+ release from
endoplasmic reticulum [14], intracellular accumulation of reactive oxygen species
(ROS) [15], mitochondrial dysfunction [16], the presence of metabolic changes,
trans-Golgi disassembly [17], and lysosomal disruption due to particulate matter
accumulation, such as alum, silica, asbestos, amyloid-
It is important to consider that NLRP3 interacts with different regulatory proteins. Many proteins, such as chaperone heat shock protein 90 (Hsp90) and its co-chaperone the suppressor of G2 allele of Skp1 (SGT1), are important for NLRP3 conformation and stability [21]. Additionally, Thioredoxin-Interacting Protein (TXNIP) serves as an oxidative sensor and interacts with thioredoxin (TRX) under reducing conditions; in the presence of ROS, TRX dissociates from TXNIP, permitting its interaction with NLRP3 and leading to inflammasome activation [22]. Moreover, guanylate-binding protein 5 (GBP5) binds to the pyrin domain of NLRP3, promoting ASC oligomerization and facilitating inflammasome activation [23]. Other factors, such as the double-stranded RNA-dependent protein kinase (PKR), migration inhibitory factor (MIF) and microtubule-affinity regulating kinase 4 (MARK4), are able to interact with NLRP3, leading to its inhibition or activation through a variety of mechanisms that involve stress sensing, autophagy, and proteasomal degradation [24, 25]. Together, these findings underscore the importance of correct NLRP3 functionality, which is involved in several cellular mechanisms responsible for intracellular degradation of exogenous stress sources, such as pathogens, and demonstrate that disruptions in these systems can contribute to tissue inflammation.
From a molecular point of view, different post-transduction modifications, such as phosphorylation, ubiquitination and SUMOylation, are involved in NLRP3 inflammasome activation and modulation, as detailed below.
NLRP3 inflammasome can be phosphorylated during both the priming phase and the
activation phase, and phosphorylation can also be involved in the inflammasome
regulation during inflammatory resolution [26]. Phosphorylation at different
sites of NLRP3 inflammasome is responsible for its activation, and several
kinases are involved in these mechanisms, including TGF-
In recent years, also SUMOylation emerged as an important post-translational modification of inflammasome. NLRP3 SUMOylation at multiple sites occurs by the E3 SUMO protein ligase MUL1 (MAPL) and regulates NLRP3 activation; in particular, it has been found that upon activation, NLRP3 becomes deSUMOylated by sentrin-specific protease 6 (SENP6) and SENP7, and defects in the SUMOylating mechanisms may lead to NLRP3 hyperactivation [37].
Since epigenetic factors, such as microRNAs and long non-coding RNA (lncRNA), have recently been recognized as crucial regulators of NLRP3 inflammasome by targeting NLRP3 mRNA [38], this section reports the epigenetic mechanisms identified as involved in NLRP3 regulation.
Among the various microRNAs implicated in NLRP3 modulation, several have been extensively characterized for their regulatory roles. For instance, a study has demonstrated that miR-9 inhibits NLRP3 inflammasome in cardiomyocytes, leading to cardiac cell death and heart failure [39]. In contrast, miR-223 inhibits NLRP3 inflammasome, resulting in reduced myocardial damage in both in vitro and in vivo models, while miR-22 acts as negative regulator of NLRP3 during tumorigenesis [40]. Similarly, miR-21 has been identified as a regulator of inflammasome activation through direct interaction with NLRP3 mRNA [41]. However, miR-21 also exerts indirect effects on NLRP3 regulation. Specifically, miR-21 targets and downregulates Phosphatase and tensin homolog (PTEN) [42], which in turn negatively regulates AKT1, a kinase responsible for NLRP3 phosphorylation [28]. This regulatory cascade suggests a complex indirect mechanism by which miR-21 influences inflammasome activity. Furthermore, Scalavino et al. [43] demonstrated that miR-369-3p reduces the expression of BRCA1/BRCA2-containing complex 3 (BRCC3), a key regulator of NLRP3 activation through de-ubiquitination [44]. By targeting BRCC3, this miRNA blocks the recruitment of ASC adaptor to the inflammasome complex, ultimately reducing NLRP3 activity.
In addition to miRNAs, several lncRNAs have emerged as crucial inflammasome regulators, modulating NLRP3 at both transcriptional and post-transcriptional level. A prominent example is Nuclear Enriched Abundant Transcript 1 (NEAT1), a lncRNA [45] normally located in the nucleus, where it is responsible for the maintenance of the structural integrity of the paraspeckles in the chromatin by associating with nuclear RNA binding proteins [46]. Upon stimulation with inflammasome-activating signals, NEAT1 disassociates from paraspeckles proteins and migrates into the cytoplasm, where it promotes inflammasome activation by facilitating the assembly of NLRP3, NLRC4 and AIM2. This occurs through NEAT1’s ability to bind and stabilize mature caspase-1 tetramers, increasing their protease activity [45]. Supporting this mechanism, an in vivo study performed on allergic rhinitis models demonstrated that NEAT1 can trigger NLRP3-mediated pyroptosis through activation of the PTBP1/FOXP1 signaling cascade [47].
Another important lncRNA in NLRP3 regulation is metastasis-associated lung adenocarcinoma transcript 1 (MALAT1). Elevated MALAT1 levels have been consistently associated with higher NLRP3 activation and consequent increased inflammation across different pathologies, including Parkinson’s disease [48], myocardial infarction [49], diabetes and atherosclerosis [50, 51, 52]. Mechanistically, MALAT1 plays a key role in the NLRP3-mediated pyroptosis in conditions such as osteoarthritis [53] and colitis [54] through its ability to modulate specific miRNAs, that regulate NLRP3 expression and activity, such as miR-124-3p [53] and miR-22-3p [54].
Together, these evidence highlight the role of NLRP3 epigenetic regulation and the disruption of these mechanisms may be involved in chronic inflammatory disorders and cancer conditions, underlining the importance of a tight and functional inflammasome regulation.
Since NLRP3 inflammasome is crucial in the modulation of phlogosis and consequently in different physiological processes several NLRP3 pharmacological modulators have been identified (Supplementary Fig. 1), which could play a role in inflammatory chronic disease treatment [55].
In neurodegenerative diseases, such as Parkinson’s disease, glibenclamide has
shown a neuroprotective effect on dopaminergic neurons, through the reduction of
over-regulated
Also hydroxybutyrate, an endogenous ketone body, showed inhibitory action on NLRP3 inflammasome probably by decreasing intracellular potassium. In a cryopyrin-associated periodic syndromes murine model, the oral delivery of exogenous hydroxybutyrate abolished inflammation [86]; in addition, in a gout murine model it blocked NLRP3 priming and activation phases in neutrophils [87]. Furthermore, inzomelid (also known as emlenoflast or MCC7840) is able to bind the NACHT site of NLRP3 and showed a successful outcome in a phase I trial for a patient with cryopyrin associated periodic syndrome [61]. Recently an innovative approach of NLRP3 modulation using an antisense oligonucleotide to target NLRP3 encoding pre-mRNA was used in mice model, resulting in NLRP3 levels reduction with consequent systematic inflammation decrease [88].
Collectively, the reported evidence highlights the importance of pharmacological NLRP3 modulation for regulating inflammatory processes in several disease conditions, and further studies are required to improve both the understanding of their molecular action and to investigate their potential clinical applications. In particular, it will be necessary to understand the clinical relevance of the reported molecules in the landscape of inflammatory diseases therapy through clinical trials, which will be essential to clarify the drug differences in efficacy and safety compared to existing treatments. This kind of studies may also be able to guide the clinical practice, determining how patient and clinical parameters, such as patient age and disease severity, may influence drug effectiveness and toxicity.
Recently emerged evidence indicates that NLRP3 pathway may be involved in the development of drug related side effects and its inhibition may represent a possible strategy to overcome drug-induced toxicity [89]. While the liver and kidney are the organs most frequently involved in adverse drug reactions, the heart, lung, skin, gastrointestinal tract, hematological system, and nervous system may also be affected by drug-induced toxicity [89]. Although the exact molecular mechanisms involving NLRP3 role in the landscape of drug-related toxicity is not completely clear, several studies have been performed confirming the importance of inflammasome in the modulation of drug adverse reactions, leading to hepatotoxicity or nephrotoxicity. This review provides a comprehensive overview of NLRP3’s involvement in drug-related side effects, with special emphasis on hepatotoxicity and nephrotoxicity, conditions affecting the primary organs responsible for drug metabolism and excretion.
Different studies demonstrated the key role of the NLRP3 inflammasome pathway in
acetaminophen-related liver injury. In particular, in vivo experiments
detected higher expression levels of both NLRP3 and IL-1
A comparable situation was identified for drug-related nephrotoxicity, which is
characterized by increased NLRP3 inflammasome levels: augmented NLRP3 expression
and consequent increase in pro-inflammatory cytokines were found in both renal
tubular and glomerular cells after cisplatin treatment in several in
vivo models [103, 104]. Zhang et al. [105] demonstrated an increased
amount of purinergic 2X7 receptor (P2X7R), involved in apoptosis and inflammation
in the renal tubular epithelial cells of mice presenting cisplatin-induced
nephrotoxicity. In contrast, pre-treatment with A-438079, an experimental
antagonist of P2X7R, significantly alleviated both cisplatin-induced renal damage
and the inflammatory response in the kidney, reducing NLRP3, ASC, caspase-1, and
ROS levels [105]. Furthermore, it was found that the caspase-1 inhibitor
quinoline-Val-Asp-difluorophenoxymethylketone (QVD-OPH) protected tubular
epithelial cells from necrosis after cisplatin treatment and was associated with
lower levels of caspase-1, IL-1
Taken together, the reported observations on both hepatotoxicity and nephrotoxicity described the possible mechanisms by which NLRP3 is modulated during drug-related adverse reactions, suggesting the importance of monitoring NLRP3 levels to predict these side effects and counteract them rapidly with different strategies, such as treating patients with NLRP3-inhibiting molecules. Also, the ability to predict the onset of drug-related side effects is fundamental to improving the outcome of pharmacological treatments related to the inflammatory status mediated by NLRP3 activity. In this context, NLRP3 methylation level detection consists of a possible biomarker to improve the anti-inflammatory precision therapy, as deeply described in the next paragraph.
The importance of NLRP3 expression and activity in the inflammatory processes
and their modulation has been thoroughly demonstrated. Indeed, the potential role
of NLRP3 as a biomarker for clinical disease management has been investigated in
various inflammatory syndromes. Currently, different inflammatory markers, such
as C-reactive protein (CRP), TNF-
In recent years, different studies have evaluated the possible impact of genetic and epigenetic factors that modulate NLRP3 levels, demonstrating their ability to predict anti-inflammatory therapy response in different disease conditions. In particular, epigenetic factors, such as DNA methylation, change with growth [116] and modulate the expression of different genes involved in drug response [117, 118]. Similarly, NLRP3 methylation levels have been investigated to evaluate their potential as biomarkers for glucocorticoid therapy response. Paugh et al. [119] identified for the first time the mechanism underlying the epigenetic regulation of the NLRP3 inflammasome in relation to glucocorticoid resistance in pediatric patients with acute lymphoblastic leukemia. In particular, the authors found that the level of promoter methylation of both NLRP3 and caspase-1 regulates glucocorticoid receptor amount, thereby modulating cell sensitivity to steroids (Fig. 2). It was shown that the overexpression of caspase-1 in a human leukemia cell line resulted in the cleavage of the glucocorticoid receptor, diminishing the glucocorticoid-induced transcriptional response and leading to higher drug resistance. The caspase-1 silencing or inhibition significantly increased glucocorticoid receptor levels, mitigating steroid resistance. They also demonstrated the fundamental role of NLRP3 in drug resistance modulation: the overexpression of CASP-1 alone, without activation via NLRP3, was not sufficient to alter leukemic cells’ sensitivity to glucocorticoids [119]. Consistently, Lucafò et al. [120] identified a correlation between NLRP3 promoter methylation levels and glucocorticoid-resistance in both adult and pediatric patients with idiopathic nephrotic syndrome. NLRP3 promoter methylation was significantly lower in steroid-resistant patients. Interestingly, on in vitro models, NLRP3 knock-down increased glucocorticoid sensitivity, whereas higher NLRP3 inflammasome activation led to a glucocorticoid receptor reduction, which was the basis of higher cellular drug resistance, thereby demonstrating a new molecular mechanism underlying steroid resistance in patients with idiopathic nephrotic syndrome [120]. The ability of NLRP3 methylation to serve as a good biomarker of glucocorticoid response was also reported in IBD patients [121]. Recent results demonstrated that steroid response is significantly associated with lower NLRP3 methylation levels in pediatric IBD patients. Moreover, the patients’ disease activity score negatively correlated with NLRP3 methylation before starting the therapy and after 1 month of methylprednisolone treatment. However, no significant association between the patients’ disease activity score or steroid response and NLRP3 methylation levels was found in adults IBD patients, whereas in those subjects, a significant positive correlation between age and NLRP3 methylation emerged, indicating that this epigenetic modification of NLRP3 changes throughout the patients’ lifespan with different clinical implications for pediatric and adult IBD forms [121].
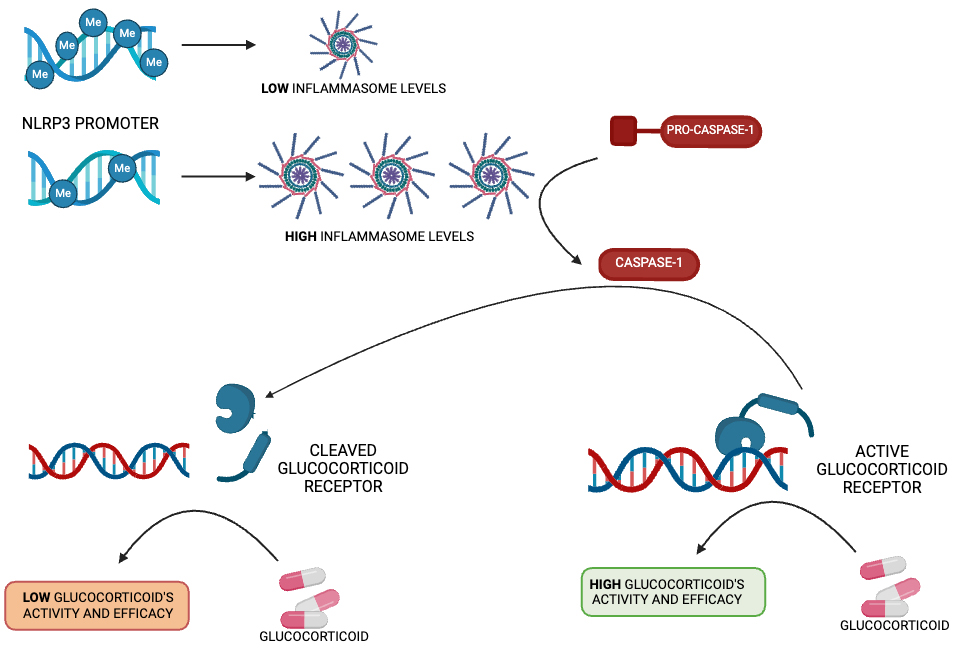 Fig. 2.
Fig. 2.
NLRP3 methylation as a glucocorticoid response biomaker. Schematic representation of the NLRP3 promoter methylation level as a regulator of glucocorticoid response.
Several recent studies have demonstrated other possible epigenetic mechanisms underlying the modulation of NLRP3 levels and affecting the drug response. Dai et al. [122] found that the lncRNA LINC00969 promotes gefitinib resistance in tumoral in vitro and in vivo models, affecting the levels of NLRP3 transcriptional m6A modification, which is an RNA post-transcriptional modification important for gene expression regulation, tumorigenesis, and drug resistance [123]. In particular, higher expression of LINC00969 in lung cancer cells with acquired gefitinib resistance could be due to the ability of this lncRNA to modulate the NLRP3 promoter transcriptionally and modify NLRP3 m6A levels. These alterations reduce inflammasome expression and block the NLRP3-mediated pyroptosis signaling pathway, promoting cancer drug resistance [122]. PTEN is a tumor suppressor that affects NLRP3 activation by directly stimulating its assembly or negatively regulating the PI3K/AKT signaling pathway, which regulates NLRP3 phosphorylation [124]. Meanwhile, BRCC3 is a deubiquitinase able to promote inflammasome phosphorylation by removing ubiquitin chains from specific lysine residues on NLRP3 [44]. Interestingly, Cheng and colleagues [125] detected that microRNA-21 blocks the expression of PTEN and BRCC3, inhibiting NLRP3 inflammasome assembly and stimulating cancer cell drug resistance to cisplatin. Furthermore, several studies have identified the epigenetic modulation of NLRP3 levels as crucial for insulin resistance [126, 127].
Together, the reported evidence highlights the crucial effect of NLRP3 level regulation, particularly by epigenetic factors, in predicting the pharmacological response to several therapies used for different pathologies presenting similar inflammatory characteristics. Indeed, further studies are required to deeply investigate the epigenetic regulation mechanisms affecting inflammasome levels to confirm the potential role of NLRP3 levels as possible pharmacological biomarkers able to predict drug efficacy.
This review thoroughly describes the crucial role of NLRP3 inflammasome in innate immune system activation and inflammation modulation. As reported, NLRP3 tight regulation is sustained by several cellular, genetic and epigenetic mechanisms, and its dysregulation is associated with several cancerous and chronic inflammatory diseases, including IBD, arthritis, gout, Alzheimer’s disease and type-2 diabetes. Several pharmacological compounds have been identified as NLRP3 modulators in vitro and in vivo models, and some of them have undergone clinical trials; however, further studies and trials are required to fully elucidate how these drugs can improve the existing therapies for inflammatory diseases both in terms of efficacy and safety.
A critical need exists for developing reliable pharmacological biomarkers that can predict patient responses to anti-inflammatory drugs. This review synthesizes emerging evidence supporting NLRP3 levels as promising candidates for personalizing therapeutic approaches, particularly for anti-inflammatory treatments that serve as primary interventions across multiple inflammatory pathologies.
Together, the collected evidence indicates that NLRP3 methylation level represent a reliable biomarker of pharmacological steroid response across multiple inflammatory disorders, highlighting its potential translatabilityacross different inflammatory pathologies that require anti-inflammatory treatments.
GZ, SS, ML, JA and GS wrote and revised the manuscript; GZ, SS, JA and ML designed the manuscript; GS and ML conceptualized the manuscript; GZ, SS, ML, JA and GS revised the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We acknowledge the European Union by the Horizon Europe PharmGenHUB Project (HORIZON-WIDERA-2021-ACCESS-02, European Commission, Grant Agreement No. 101059870).
This work was supported by the Italian Ministry of Health, through the contribution given to the Institute for Maternal and Child Health IRCCS Burlo Garofolo, Trieste, Italy, (grant numbers: RC 15/23).
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBL39537.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.