
- Academic Editor
Systemic lupus erythematosus (SLE) is a chronic autoimmune disorder characterized by immune system dysfunction, the production of autoantibodies, and multi-organ inflammation. Lupus nephritis (LN) is one of the most severe complications of this condition. Approximately 60% of patients with SLE develop LN, which leads to both increased morbidity and mortality. Furthermore, LN has the potential to progress to end-stage renal disease. Macrophages, key components of the innate immune system, are involved in the pathophysiology of LN through immune complex clearance, antigen presentation, regulation of inflammation, and tissue repair. Macrophage polarization into pro-inflammatory (M1) versus anti-inflammatory (M2) functional phenotypes is a component of LN disease progression. M1 macrophages are responsible for supporting pro-inflammatory immunity and promoting tissue damage, whereas M2 macrophages are necessary for tissue repair and resolution of inflammation. However, dysregulated M2 function may exacerbate the pathogenesis of LN, indicating the complex role of macrophages in LN. Novel therapeutic approaches associated with the mechanisms of macrophage polarization and/or macrophage signaling pathways have emerged as therapeutic targets to modify the progression of LN. Furthermore, proinflammatory cytokines enhance renal inflammation and autoimmunity; alternatively, anti-inflammatory cytokines play a dual role in LN, contributing positively and negatively to the disease process. The purpose of this review is to investigate the role of macrophages in the pathogenesis of LN and highlight macrophage-targeted therapies or biomarkers as diagnostic tools and new therapeutic avenues to improve long-term outcomes for patients.
Systemic lupus erythematosus (SLE) is a chronic and systemic autoimmune disease where the immune system attacks its own tissues and organs, leading to the production of autoantibodies and immune complexes that cause widespread inflammation and damage to multiple organ systems [1, 2, 3, 4, 5]. The term “lupus”, the Latin word for “wolf”, refers to the historical identification of a facial rash resembling a wolf bite, one of the clinical manifestations of this disease [6, 7, 8]. However, SLE is not limited to the skin but can affect almost every organ system in the body including the musculoskeletal, renal, cardiac, pulmonary, and central nervous systems [4, 9]. The disease is highly heterogeneous and is characterized by a range of symptoms, including fatigue, fever, arthralgia, skin rashes, and renal involvement, contributing to SLE being one of the most complicated autoimmune disorders to diagnose and treat [4, 9]. SLE is more prevalent in women of childbearing age, and is also more common in non-Caucasians including those of African American, Asian, and Hispanic descent [10]. The etiology of SLE remains unclear but is thought to be a combination of genetic, environmental (ultraviolet light, infections, and certain medications), and hormonal etiologies [2, 4].
Lupus nephritis (LN) is one of the most severe complications of SLE, affecting up to 60% of patients with SLE during the disease course, leading to increased morbidity and mortality [11, 12]. In addition, approximately 7.9% of patients diagnosed with LN progress to end-stage renal disease within 16.5 years of LN onset [4, 5, 11, 12]. The poor prognosis of LN highlights the crucial need for more diagnostic strategies and effective United States Food and Drug Administration-approved therapies to slow kidney damage and reduce morbidity and mortality. The cellular processes involved in the development of LN diagnosis are multifactorial, involving immune mechanisms including immune complex deposition, impaired clearance of apoptotic cells, and loss of tolerance to autoantigens, while abnormal immune activation is also a contributing factor [2, 4, 13]. Macrophages, which are integral components of the innate immune system, play a multifaceted role in LN including clearing immune complexes, presenting antigens, regulating inflammation, and participating in tissue repair. Their complex role makes them central to imaging immune cell processes in LN [14]. The polarization of macrophages also plays a substantial role in determining disease progression, with pro-inflammatory (M1) macrophages associated with driving inflammation, tissue damage, and injury, while anti-inflammatory (M2) macrophages promote tissue repair and restoration of inflammation back to basal levels. The prominent role of macrophages in LN suggests that therapies targeting macrophage polarization and signaling could be promising therapeutic strategies [14, 15, 16]. The increasing understanding of the cellular and molecular processes of macrophage polarization and activation will lead to a surge in the applicability of therapeutic approaches targeting macrophage signaling. Furthermore, biomarker identification may also play a role in clinical translation, aiding in early diagnosis, disease monitoring, and treatment paradigms in LN. Understanding the pathogenesis of LN, particularly the role of macrophages, is crucial for advancing therapeutics aimed at modulating organ damage and subsequent inflammation, and promoting healthy immune responses. This review provides a thorough overview of the role of macrophages in LN, discussing emerging therapeutic strategies that may improve patient outcomes.
The polarization of macrophages plays an important yet paradoxical role in the pathogenesis of LN, reflecting the dynamic balance between pro-inflammatory and anti-inflammatory macrophage phenotypes. In the context of LN, macrophages exhibit remarkable plasticity in their ability to switch between M1-like (classically activated) and M2-like (alternatively activated) states in response to environmental cues (Fig. 1). This macrophage plasticity is often disrupted in autoimmune conditions and contributes to a worsening LN phenotype. Thus, maintaining a balance between these phenotypes is essential for overall immune system homeostasis, and its disruption can result in chronic inflammation and tissue damage.
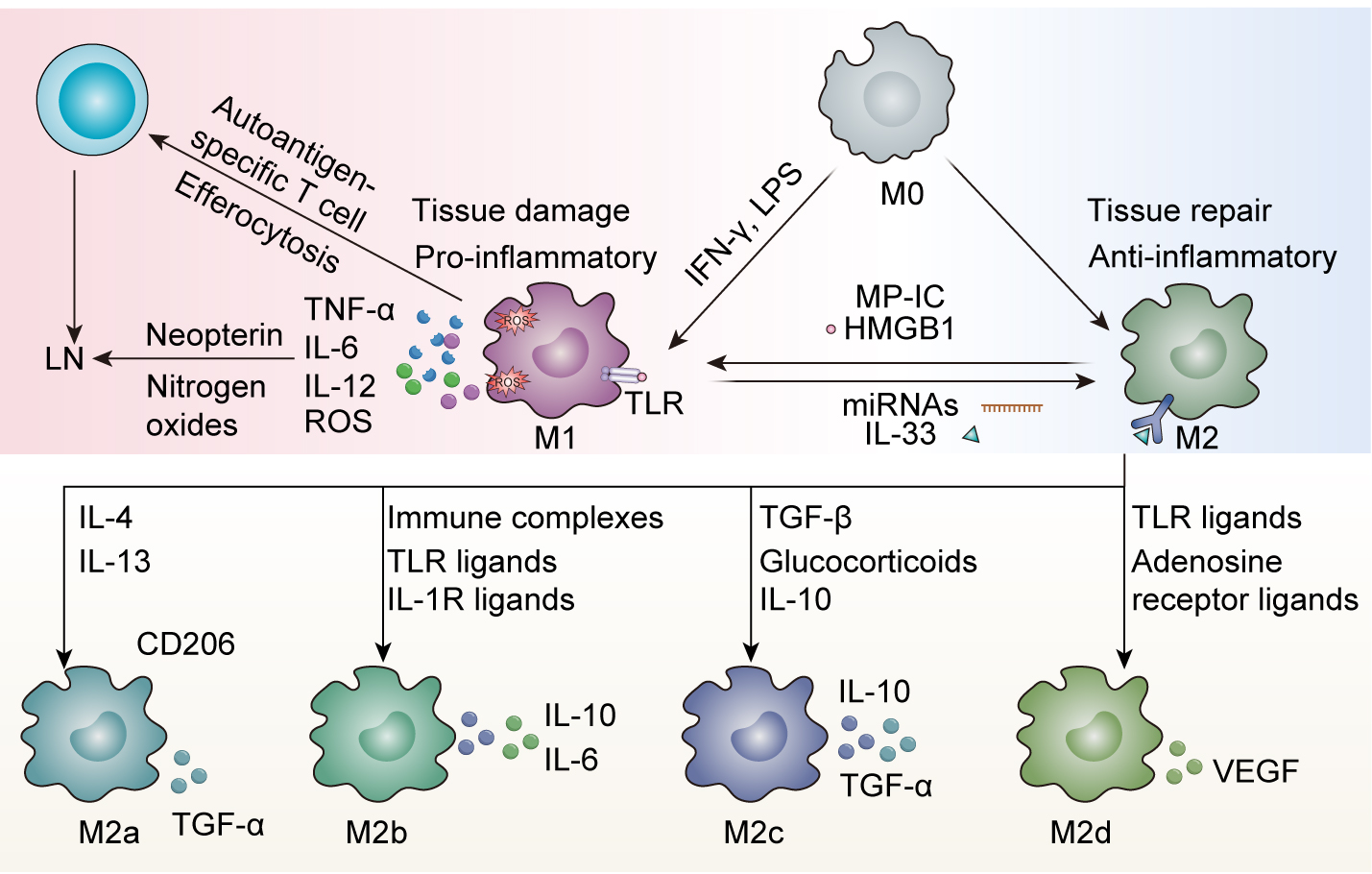 Fig. 1.
Fig. 1.
Macrophage plasticity in Lupus nephritis (LN). Macrophage
polarization represents a dynamic equilibrium between pro-inflammatory (M1) and
anti-inflammatory (M2) phenotypes that critically influences LN pathogenesis. M1
macrophages, activated by T helper 1 (Th1) cytokines (e.g., interferon gamma
(IFN-
M1 macrophages are generally prominent in acute inflammation, and are
classically activated by T helper 1 (Th1) cytokines such as interferon gamma
(IFN-
M2 macrophages are known for their anti-inflammatory, tissue-repair, and
angiogenesis-promoting roles. They are induced by agents such as Th2 cytokines
(IL-4, IL-13), IL-10, and glucocorticoids, and also secrete factors like IL-10
and transforming growth factor beta (TGF-
Importantly, while these classifications exist, the accurate in vivo translation of M2 subtypes is difficult. This suggests that M2 classification is reflective of the stimuli for macrophage activation instead of the functions that occur after activation. Additionally, the functional attributes of macrophages are not determined by their origin, but are instead modified by the local microenvironment, indicating that macrophage polarization is dynamic and context-dependent. This nuance underscores the need for targeted therapies that modulate macrophage function in disease, especially LN, where modulation of macrophage function is essential for disease improvement. In the context of their involvement in LN pathology, M2-polarized macrophages contribute to the resolution of inflammation. However, sometimes defective M2 function, as seen with decreased heme oxygenase-1 expression, leads to LN progression due to the insufficient clearance of immune complexes, and can perpetuate the production of pro-inflammatory signaling [31]. In some cases, certain M2 subtypes, including M2b, can worsen disease by secreting both pro-inflammatory and anti-inflammatory cytokines [21, 32, 33]. One particularly interesting finding is that the use of adoptively transferred M2 macrophages reduce disease severity in murine models of LN, indicating their potential therapeutic benefit if properly regulated [34]. The ability of M2 macrophages to influence the immune response and promote tissue repair makes them an attractive therapeutic target in LN.
The dynamics of macrophage polarization in LN are deeply influenced by a variety of microenvironmental factors that disturb the balance between pro-inflammatory M1 and anti-inflammatory M2 phenotypes. High-mobility group box 1 (HMGB1), which is a damage-associated molecular pattern, induces M1 polarization and worsens LN severity by enhancing antigen presentation and T-cell activation [21, 34, 35]. HMGB1 is released from necrotic and activated immune cells and binds to TLRs and the receptor for advanced glycation end products to further exacerbate the inflammatory response [35]. Microparticle-bound immune complexes (MP-ICs) are released from apoptotic cells and carry autoantigens that lead macrophages to polarize to an M1-like phenotype, which further promotes inflammation in SLE [36]. By contrast, IL-33 is an alarmin cytokine released by damaged epithelial cells, which promotes M2 polarization and regulatory T cell (Treg) expansion through ST2 receptor signaling [37]. These processes, in turn, mitigate the severity of LN in preclinical models by promoting tissue “reparative” actions and anti-inflammatory functions. MicroRNAs (miRNAs), including miR-150, play a crucial role in regulating macrophage polarization by targeting transcription factors such as suppressor of cytokine signaling 1 (SOCS1) [38]. Dysregulation of miRNAs has been linked to fibrosis and immune dysregulation associated with LN, as miRNAs can regulate gene expression at the post-transcriptional level and contribute to excessive inflammatory responses [39]. Collectively, these factors reveal the complex interplay of molecular signals that regulate macrophage polarization and their significant contribution to both the pathogenesis and progression of LN.
The immunological features of LN involve immune complex deposition in the glomeruli, leading to nephritis, progressive kidney injury, and ultimately renal failure if it is not treated. Macrophages, which are vital components of the innate immune system, are fundamentally important in the pathophysiology of LN, as they contribute to both inflammation and subsequent fibrotic processes that drive disease progression. An elaborate network of cytokines and chemokines regulate macrophage activation, polarization, and recruitment in LN—all of which coordinate the immune response and the remodeling of tissue. Defining and understanding the role of these mediators is crucial for designing strategies to selectively alter renal inflammation and fibrosis in LN.
Pro-inflammatory cytokines are key mediators of renal inflammation in LN that
facilitate the recruitment and activation of immune cells and production of
pathogenic autoantibodies and immune complexes, which are subsequently deposited
in renal tissue (Table 1). TNF-
| Category | Key mediators | Source | Role in LN pathogenesis | Therapeutic implications |
| Pro-inflammatory cytokines | TNF- |
M1 macrophages | Increases adhesion proteins (ICAM-1, VCAM-1) | Inhibition reduces glomerular hypercellularity and proteinuria in preclinical models |
| Promotes leukocyte infiltration | ||||
| IL-6 | Macrophages, B cells | Promotes B-cell differentiation | Targeting IL-6 signaling alleviates renal inflammation | |
| Enhances monocyte and neutrophil recruitment | ||||
| IL-1 |
Macrophages (inflammasome) | Drives MC activation Enhances glomerulosclerosis | Inhibition reduces inflammatory infiltrates and improves renal function | |
| IFN- |
Th1 cells, M1 macrophages | Increases MHC class II expression | Contributes to autoantigen presentation and immune cell recruitment | |
| Sustains chronic inflammation | ||||
| IL-12 | Macrophages, DCs | Promotes Th1 polarization | Drives renal parenchyma injury | |
| Enhances killing function of natural killer cells | ||||
| Anti-inflammatory cytokines | IL-10 | M2 macrophages | Inhibits TNF- |
Dual role: protective (limits inflammation) and pathogenic (promotes autoimmunity) |
| Promotes B cell survival and autoantibody production | ||||
| TGF- |
M2 macrophages | Promotes tissue repair | High levels correlate with disease activity and fibrosis | |
| Contributes to fibrosis | ||||
| IL-4 | Th2 cells, M2 macrophages | Promotes Th2 differentiation | Limits inflammation but may contribute to fibrosis | |
| Activates M2 macrophages | ||||
| Chemokines | CCL2 (MCP-1) | Renal endothelial/epithelial cells | Recruits monocytes and macrophages via CCR2 | Blocking CCL2/CCR2 reduces macrophage infiltration and improves renal function |
| Sustains renal inflammation | ||||
| CX3CL1 (Fractalkine) | Glomeruli, renal arteries | Recruits monocytes and macrophages via CX3CR1 | Inhibition reduces macrophage numbers and inflammation | |
| Localized role in renal inflammation | ||||
| CCL3/CCL4/CCL5 | Macrophages, T cells | Recruits immune cells via CCR1/CCR5 | CCR1/CCR5 signaling suppression reduces inflammation | |
| Drives immune infiltrate into the kidney | ||||
| CXCL9/CXCL10 | Renal tubular epithelial cells | Recruits CXCR3+ T cells and macrophages | Blocking CXCL9/CXCL10 may limit immune cell recruitment and inflammation | |
| Upregulated by IFN- |
ICAM-1, intercellular adhesion molecule-1; VCAM-1, vascular cell adhesion
molecule-1; DCs, dendritic cells; MC, mesangial cell; MHC, major
histocompatibility complex; CCL2, C-C motif ligand 2; MCP-1, monocyte
chemoattractant protein-1; CCR2, C-C chemokine receptor type 2; CX3CL1, C-X3-C
motif ligand 1; LN, Lupus nephritis; TNF-α, tumor
necrosis factor alpha; IFN-
Anti-inflammatory cytokines such as IL-10, TGF-
Chemokines, small signaling proteins that are critical to activating immune
cells, contribute to the pathogenesis of LN by recruiting monocytes, macrophages,
and any immune cell type into the kidney that can promote inflammation and tissue
injury (Table 1). Among the chemokines studied, CCL2 and its receptor, C-C
chemokine receptor type 2 (CCR2), have garnered the most research attention. CCL2
is produced by renal endothelial and epithelial cells in response to inflammatory
stimulation, and acts as a chemoattractant for monocytes and macrophages, which
bind to CCR2 and are subsequently recruited to the kidney [52]. Immune cell
recruitment is important for both initiating renal inflammation and sustaining
renal inflammation. In the lupus-prone mouse models, MRL/MpJ strain with the
lymphoproliferation mutation in the Fas gene (MRL/lpr) and New Zealand
Black
The interplay of cytokines and chemokines significantly contributes to the
complex networks that drive the pathogenesis and progression of LN. For example,
mesangial-derived IL-6 can induce CCL2 expression in macrophages, which recruits
additional monocytes to the kidney, leading to inflammation [71]. TNF-
Macrophages serve as important coordinators of the immune response in LN, establishing complex interactions with both immune and non-immune cells that mediate disease progression (Fig. 2).
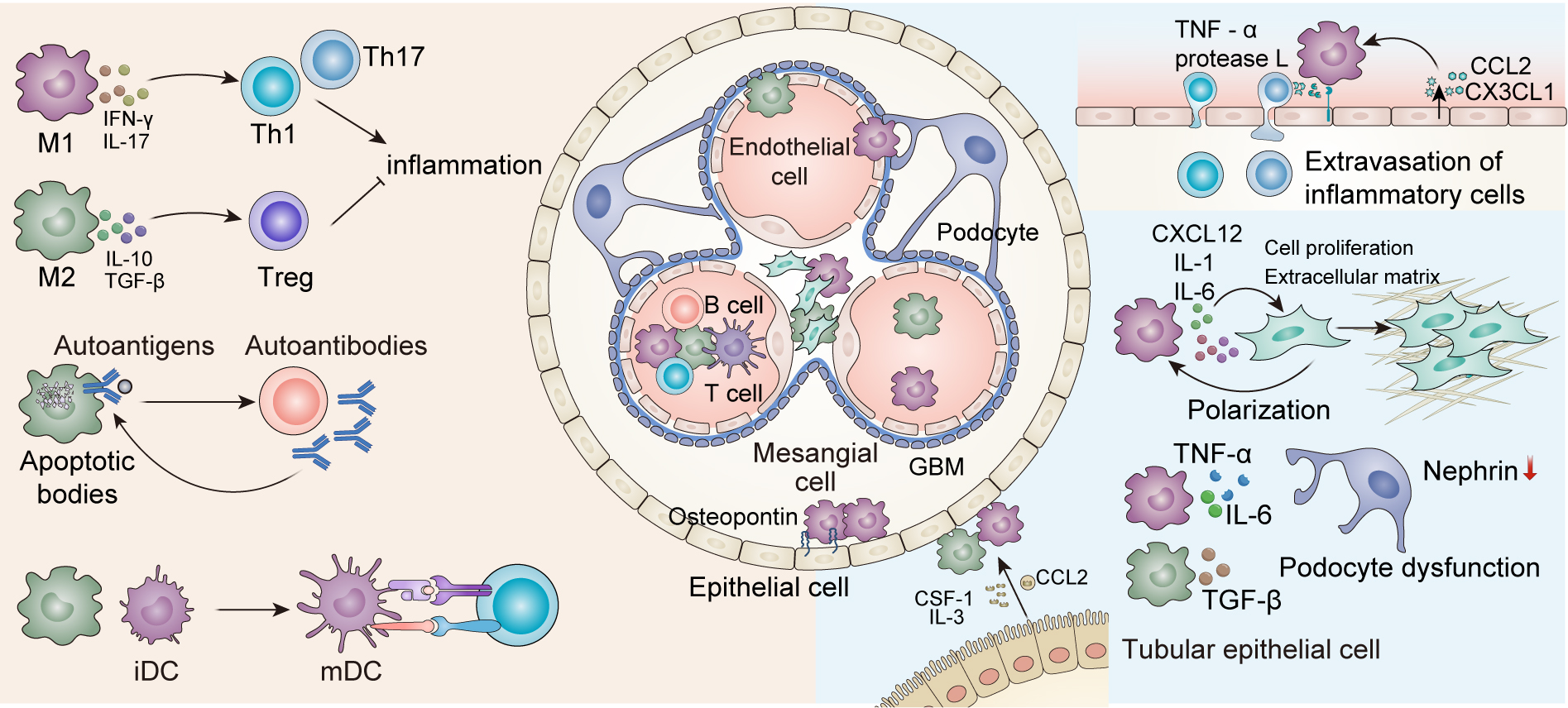 Fig. 2.
Fig. 2.
Interactions of Macrophages with other cells in LN. Within the
adaptive immune system, macrophages engage in bidirectional crosstalk with T
cells, influencing T-cell polarization. M1-polarized macrophages promote Th1/Th17
immune responses via IFN-
In the immune compartment, macrophages interact with T cells, B cells, and dendritic cells (DCs) to modulate adaptive immune responses by promoting either Th1, Th17, or Treg responses, depending on the macrophage polarization. In turn, the interactions among T cells, B cells, and DCs determine whether macrophages produce pro-inflammatory or anti-inflammatory cytokines, creating the inflammatory environment in LN.
M1-like macrophages play a crucial role in driving amplified Th1 and Th17
responses, which lead to increased injury and inflammation via inflammatory
cytokines such as IFN-
In addition to their dialogue with immune cells, macrophages also interact with various non-immune renal cells, including glomerular endothelial cells, MCs, podocytes, and renal tubular epithelial cells, contributing to renal tissue injury, fibrosis, and dysfunction. Macrophages, through the secretion of cytokines, chemokines, and matrix-degrading enzymes, exacerbate inflammation and disrupt the structural integrity of the kidney; in turn, renal cells subsequently regulate the recruitment, polarization, and survival of these macrophages.
Macrophages release pro-inflammatory cytokines, including TNF-
These various cell interactions highlight the multifaceted role of macrophages in LN, which drives and modulates inflammation and tissue damage by directly interacting in a complex manner with a variety of cell types. The reciprocal crosstalk between macrophages and immune and non-immune cells highlights their central role in LN pathogenesis and demonstrates the importance of developing therapeutic strategies to target their interactions.
Macrophages play a critical role in the development of LN and are therefore a promising target for therapeutic interventions (Table 2). As mediators of inflammation, kidney injury, and fibrosis, macrophages also interact with immune and non-immune cells in the kidney. Strategies that could be utilized to manipulate macrophages include targeting the pro-inflammatory actions of macrophages, enhancing the actions of anti-inflammatory or reparative macrophages, and/or blocking the ability of macrophages to migrate to the kidney. Targeting macrophages is a new and exciting approach for managing LN that holds promise for improving outcomes in this complicated and burdensome disease.
| Strategy | Mechanism | Key targets/agents | Effects | Challenges |
| Enhancing phagocytic efficiency | Boosting macrophage clearance of apoptotic cells & immune complexes | - EPO signaling (ARA290, non-erythropoietic EPO derivative) | - Reduces autoantibodies & renal inflammation | - EPO agonists cause hypertension/thromboembolism |
| - PPAR |
- Shifts macrophages to anti-inflammatory phenotype | - Need optimized PPAR | ||
| - Efferocytosis enhancers (Hv1, serum amyloid P component, milk fat globule epidermal growth factor 8, growth arrest-specific gene 6) | ||||
| Blocking macrophage recruitment | Inhibiting chemokine-driven infiltration into kidneys | - CCL2/CX3CL1 inhibitors (Mizoribine, Disulfiram) | - Reduces renal macrophage infiltration | - Long-term safety of chemokine blockade unclear |
| - CSF-1R inhibitor (GW2580) | - Lowers proteinuria & chronicity index | |||
| - TLR7/9 inhibitors (dexamethasone nanogels) | ||||
| Shifting polarization | Promoting anti-inflammatory M2 macrophages | - IL-33/ST2 pathway | - Decreases pro-inflammatory cytokines (TNF- |
- Risk of over-suppressing immune responses |
| - Natural compounds (paeonol) | - Improves renal function | - Notch pathway modulation complexities | ||
| - MSCs | ||||
| - LXR |
||||
| - Azithromycin (PI3K/Akt pathway) | ||||
| Suppressing cytokine secretion | Inhibiting pro-inflammatory cytokine release | - NLRP3 inhibitors (MCC950) | - Reduces IL-1 |
- Glucocorticoid side effects |
| - Natural compounds (paeoniflorin, luteolin) | - Attenuates NF-κB activation | - Need for targeted delivery | ||
| - HMGB1 antagonists | ||||
| - MSC-derived exosomes (miR-146a-5p, miR-16) | ||||
| Reprogramming metabolism | Shifting glycolysis (M1) → oxidative phosphorylation (M2) | - Glycolysis inhibitors | - Reduces IL-1 |
- Metabolic pathways are complex & interconnected |
| - CPT1a enhancers (promotion of fatty acid oxidation) | - Enhances tissue repair | |||
| - mTOR modulators | ||||
| - PPAR |
||||
| miRNA-based therapy | Regulating macrophage polarization & inflammation via miRNAs | - miR-181d-5p antagonists | - Suppresses pyroptosis & NLRP3 activation | - Delivery challenges |
| - MSC exosomes (miR-13896, miR-146a-5p) | - Promotes M2 polarization | - Off-target effects possible | ||
| - miR-155/miR-21 inhibitors |
EPO, erythropoietin; PPAR
Macrophages are essential for the clearance of apoptotic cells and immune
complexes, both of which are thought to be involved in the pathogenesis of LN.
Augmenting the phagocytic activity of macrophages has been proposed as a
therapeutic strategy to reduce renal inflammation and preserve renal function in
LN. One approach is to target erythropoietin (EPO) signaling, which is a critical
mediator of the phagocytic clearance of apoptotic cells. In lupus mouse models,
EPO has been shown to decrease autoantibody production and improve renal
function, partly by enhancing macrophage phagocytic activity [90]. However,
the ability to clinically utilize EPO agonists is limited by adverse effects,
particularly hypertension and thromboembolism [91]. To address some of these
challenges, a non-erythropoietic EPO derivative of ARA290 is being investigated.
ARA290 also enhances macrophage phagocytosis of apoptotic cells, inhibits the
differentiation of pro-inflammatory M1 macrophages, and inhibits the production
of pro-inflammatory cytokines such as IL-6 and TNF-
An additional and alternative therapeutic strategy is targeting peroxisome
proliferator-activated receptor gamma (PPAR
Efferocytosis (the process of macrophages clearing apoptotic cells) is inhibited in LN by the continuing deposition of autoantigens, which allows for the development and persistence of autoimmunity, thereby inhibiting efferocytosis. Several researchers have been interested in the restoration of efferocytosis as a possible therapeutic approach to limit inflammation in the kidney and improve renal function in LN. One potential target is voltage-gated proton channel 1 (Hv1), which is required for acidification of the phagosome for efferocytosis. Increased Hv1 activity promotes the clearance of apoptotic cells and decreases autoantibody production in lupus-prone mouse models [97]. The serum amyloid P component shifts macrophage polarization from the pro-inflammatory M2b phenotype to the anti-inflammatory M2a phenotype, thus promoting efferocytosis and reducing the severity of LN [98, 99]. Additionally, molecules such as milk fat globule epidermal growth factor 8 and growth arrest-specific gene 6 can facilitate the removal of apoptotic cells (efferocytosis) by binding to phosphatidylserine [100, 101]. These mechanisms provide breakthroughs in restoring efferocytosis to help limit inflammation and improve renal function in LN.
Targeting macrophage recruitment to the kidneys is a promising therapeutic
approach in LN, as macrophages play a key role in kidney inflammation and tissue
damage. The recruitment of macrophages is mediated through chemokine signaling,
adhesion molecules, and TLR activation, each of which has been targeted in
preclinical and clinical studies. For example, the chemokines CCL2 and CX3CL1 are
upregulated in LN, promoting the recruitment of inflammatory cells to the kidney
[46]. Mizoribine, a purine synthesis inhibitor, reduces macrophage infiltration
into the kidney by downregulating OPN, a key chemokine in monocyte recruitment.
Mizoribine has shown efficacy in clinical studies in preventing macrophage
infiltration and reducing the chronicity index in patients with LN [102, 103, 104, 105, 106].
Disulfiram, an alcohol withdrawal drug, inhibits monocyte/macrophage migration
via FROUNT, a cytoplasmic protein that promotes chemotaxis via CCR2 and
CCR5 and suppresses pro-inflammatory elements such as TNF-
The polarization of macrophages is a significant factor in the pathogenesis of
LN, as the balance of pro-inflammatory M1-like and anti-inflammatory M2-like
forms is part of the disease pathogenesis and resolution. The characterization of
pathways, mechanisms, and molecular regulators involved in macrophage
polarization has broadened the therapeutic landscape for LN. IL-33 is one of
these regulators and has been defined as one of the strongest potential
macrophage polarization regulators discovered to date. IL-33 promotes M2
macrophage polarization and Treg proliferation by upregulating ST2, leading to
the autoregulation of M2 macrophages and Tregs [115, 116, 117]. Preclinical studies in
mouse models in LN have shown that treatment with IL-33 leads to decreased renal
inflammation and proteinuria, indicating that IL-33 may be a promising
therapeutic target in LN [115, 116]. Similarly, another therapeutic approach using
natural compounds (e.g., paeonol) induces M2 polarization of macrophages through
inhibition of mitogen-activated protein kinase and nuclear factor kappa B
(NF-
Endoplasmic reticulum stress (ERS) is increasingly understood to be a
fundamental driver of LN pathobiology through the activation and polarization of
macrophages. ERS is considered the initial event leading to the unfolded protein
response, a cellular stress response that restores protein homeostasis. Prolonged
ERS leads to the production of pro-inflammatory cytokines and NLRP3 inflammasome
activation, both of which promote renal inflammation and injury in LN [123].
Therefore, modulation of ERS pathways is an attractive therapeutic strategy to
reduce inflammatory responses, potentially mediated by macrophages, and restore
renal function. Advances in drug development and structure-based drug design have
resulted in the development of selective small molecule inhibitors and drugs that
can be assessed in preclinical and clinical trials. For example, several
inhibitors of IRE1
In LN, pro-inflammatory cytokines released from macrophages are a leading
contributor to renal inflammation and damage, making inhibiting cytokine release
a reasonable therapeutic target. Glucocorticoids are one of the most frequently
employed treatments for LN due to their strong anti-inflammatory effects by
decreasing secretion of IL-1
Macrophage metabolism is integral to defining their functional phenotype,
particularly in the LN context. Metabolic reprogramming has been identified as a
potential therapeutic approach. M2 macrophages rely on oxidative phosphorylation,
a metabolic route that is bioenergetically favorable and allows M2 macrophage
involvement in tissue repair functions [137]. One study demonstrated that
inhibiting glycolysis can reduce kidney inflammation by decreasing the release of
IL-1
MiRNAs serve as key regulators of macrophage function in LN and are promising targets for therapeutic approaches, as they regulate gene expression at the post-transcriptional level. These small non-coding RNAs can significantly impact macrophage polarization and the inflammatory response to injury, and are well-established contributors to the pathogenesis of LN. For example, miR-181d-5p is an important contributor to renal inflammation and pyroptosis [147, 148]. In exosomes that have been released from macrophages, increased expression of miR-181d-5p is a mechanism for targeting B-cell lymphoma 2 (anti-apoptotic protein) and regulating the NLRP3/caspase-1/gasdermin D pathway responsible for inducing pyroptosis [148]. Furthermore, antagonism of miR-181d-5p has been shown to diminish renal damage and inflammation, indicating that exosomal miRNAs represent potential therapeutic targets in LN [147]. Thus, the concept of utilizing miRNA therapy to treat LN by reducing inflammation mediated by macrophages is promising, specifically mi-R181d-5p and miR-13896 packaged in exosomes from MSCs, which have shown favorable therapeutic outcomes. Similar to miR-181d-5p, exosomes can mediate M2 macrophage polarization and enhance tissue repair functions through mechanisms that block macrophage activity. The miR-13896 exosomes have been shown to improve renal function and reduce renal inflammation. The use of exosomes derived from MSCs to improve LN appears promising [149]. In addition, exosomes derived from MSCs that contain miR-146a-5p and miR-16 also provide protective effects through different pathways, with miR-146a-5p downregulating the Notch 1 signaling pathway, which is associated with proliferation and inflammation [150]. The miR-16 components promote M2 macrophage polarization. Collectively, they provide a method to diminish collagen deposition in the glomeruli and decrease complement C3 levels, while promoting renal function. Other than exosome-packaged miRNAs, miR-21 and miR-155 are also related to LN pathology [151]. While miR-21 can promote fibrosis and inflammation, miR-155 can promote M1 macrophage differentiation and enhance the release of inflammatory cytokines. Thus, inhibiting miR-155 and miR-21 may lead to a shift in cytokine production towards a more anti-inflammatory M2 phenotype, ultimately leading to less inflammation and improved renal function in patients with LN. This information demonstrates the multifaceted roles of miRNAs in modulating the activity of macrophages and their potential as therapeutic targets. Thus, miRNA therapeutics that target specific mRNAs and modulate macrophage activity and renal inflammation in LN may be a novel and effective treatment in the future.
Identifying reliable biomarkers for LN that will aid in early diagnosis, monitor disease activity, and identify personalized treatment is of great value. Some of the discoveries made in molecular and cellular studies in recent years have revealed numerous informative biomarkers for disease activity, renal involvement, and treatment response. These biomarkers provide some insights into the underlying pathophysiology of LN, and in some cases therapeutic targets (Table 3).
| Category | Key biomarkers | Clinical utility | Therapeutic implications |
| Cell-based biomarkers | CD11c+ macrophages in urine | Proliferative LN diagnosis | Predictive models for therapy response |
| scRNA-seq-defined macrophage subsets | Monitoring treatment response | Targeting macrophage polarization | |
| Cytokine/Chemokine | M-CSF, IL-6, TNF- |
Disease activity monitoring | Anti-cytokine therapies (e.g., anti-IL-6, anti-TNF- |
| Renal involvement assessment | |||
| Genetic/Epigenetic | Hypomethylation (IFN-induced protein with tetratricopeptide repeats 1, MX dynamin-like GTPase 1) | Risk stratification | Type I IFN inhibitors (for IFN-high patients) |
| miR-150 upregulation | Renal damage progression | B-cell depletion (for plasma blast-high) | |
| Neutrophil/plasma blast gene signatures | |||
| Urinary biomarkers | sCD163 (macrophage activation) | Early LN diagnosis | Tubular repair strategies (EGF) |
| EGF (↓ = poor prognosis) | Chronic kidney disease progression prediction | Anti-fibrotic therapies (uPAR) | |
| uPAR (↑ = progression) | |||
| Clinical/Histological integration | Proteinuria ( |
Long-term remission prediction | - |
| Tubulointerstitial inflammation/chronicity index | Renal prognosis |
EGF, epidermal growth factor; uPAR, urokinase-type plasminogen activator receptor.
In the pathophysiology of LN, macrophages, particularly their polarized states (M1 and M2), contribute to both progression and regression. CD11c+ macrophages in urine have also been identified as potential biomarkers of proliferative LN [152, 153]. These urinary CD11c+ macrophages originate from circulating monocytes, and are found in increased numbers in the urine of patients with active proliferative LN. They are significantly decreased in patients with complete or partial renal response to therapy. The presence of CD11c+ macrophages is associated with elevated serum anti-dsDNA antibody levels, renal tubular atrophy, and interstitial fibrosis, all of which suggest the utility of this cell type as a marker of LN. Single-cell RNA sequencing (scRNA-seq) is a powerful tool for studying cellular heterogeneity in LN by providing unprecedented granularity in gene expression analysis at the single-cell level [154]. Specific macrophage subsets have emerged as cellular contributors to renal injury. The interplay of these subsets has led to the development of predictive models to assess individual patient responses to therapies. ScRNA-seq studies have revealed that tissue-resident macrophages exhibit a diverse range of transcriptional profiles, including both IFN and mixed anti-inflammatory signatures [155]. These data demonstrate the potential of scRNA-seq to discover novel markers and identify predictive targets for therapy.
Cytokines and chemokines are produced by macrophages or other immune cells may also
be involved in the pathogenesis of LN. Macrophage CSF (M-CSF), predominantly
produced by circulating monocytes, is elevated in patients with lupus and linked
to both disease activity and renal involvement [156]. Furthermore, M-CSF is
positively correlated with the number of circulating monocytes and is likely
associated with the recruitment and activation of macrophages within the kidney
[157]. Elevated levels of cytokines like IL-6, TNF-
The presence of both genetic and epigenetic factors adds to the heterogeneity of
LN and may also serve as a biomarker for risk prediction and disease progression.
The hypomethylation of genes involved in the type I IFN response has been shown
in the immune cells of patients with SLE. Profiling the epigenetic landscape may
help identify patients at higher risk of LN. The hypomethylation of genes such as
IFN-induced protein with tetratricopeptide repeats 1 and MX dynamin-like GTPase 1
correlate with increased disease activity and renal involvement [158]. miR-150 is
another example of a miRNA that is upregulated in patients with LN and is
involved in both fibrosis and inflammation, correlating with the severity of
renal damage. miR-150 promotes renal fibrosis through the downregulation of
SOCS1, a negative regulator of NF-
Urinary biomarkers are emerging as potential predictors of treatment response. Soluble CD163 (sCD163), a marker of macrophage activation, has emerged as an important biomarker in LN. In urinalysis, elevated sCD163 levels are a strong indicator of active LN and correlate with disease activity, histopathology, and treatment response [164]. sCD163 levels may begin to increase up to 6 months before an individual develops LN, suggesting its utility as a biomarker for early diagnosis. Proteomic analysis of urine will identify panels of biomarkers associated with renal outcomes. As an example, urinary epidermal growth factor (EGF) and soluble urokinase-type plasminogen activator receptor (uPAR) are independent predictors of chronic kidney disease progression in LN [165, 166, 167]. EGF plays a role in tubular repair/regeneration and decreased levels are associated with poorer renal outcomes. Similarly, uPAR is an inflammatory and fibrotic marker, and elevated levels predict disease progression. Incorporating these urinary biomarkers into clinicopathologic predictive models will improve the accuracy of predicting treatment response.
The establishment of models predicting response to treatment plays an important
role in a multitude of factors and approaches considered in treatment
decision-making in LN. A better understanding of the clinical patient profile
(e.g., class, duration of disease, and onset of LN), clinical characteristics
(e.g., proteinuria, serum creatinine), and histologic characteristics (e.g.,
tubulointerstitial inflammation, index of chronicity) can lead to improved
individualized therapy and effective treatment plans. It also provides avenues
where predictive models can leverage clinical data, such as laboratory results,
urine protein measures, and histopathological findings to enhance treatment
response prediction. For instance, tubulointerstitial inflammatory cell
infiltrates and a high chronicity index indicate worse renal outcomes [168].
Additionally, a proteinuria level of
The study of macrophage polarization has revealed possible avenues for future research and therapeutic development, especially as we learn more about the complex functions of M1 and M2 macrophages in the pathophysiology of LN. One key area of interest is selective modulation of the macrophage polarization state to restore the balance between the pro-inflammatory M1 phenotype and anti-inflammatory M2 phenotype. Recently, there has been interested in developing therapeutics that target specific macrophage signaling cascades, including those involving molecules like HMGB1, IL-33, and miRNAs. The development and application of accurate biomarker assessment for macrophage polarization states, such as CD163, CD206, and specific cytokines, would be useful for early diagnostic and follow-up assessments of LN if the biomarkers could be assessed through non-invasive methods such as urine or blood samples. Lastly, we should look beyond the modulation of cytokines in macrophage polarization and consider examining novel potential therapeutic targets in the macrophage signaling network as well as the roles of extracellular vesicles and microparticles in macrophage polarization and the progression of LN. The M1/M2 classification is an oversimplification of macrophage behavior in LN, as real-world macrophages exhibit hybrid phenotypes and dynamic plasticity. Murine studies are limited by species-specific differences, whereas human LN involves IFN-driven and metabolic influences. Spatial heterogeneity and unreliable biomarkers further complicate the M1/M2 framework. Future research should integrate advanced models (organoids, spatial omics) and target specific dysfunctional pathways rather than broad polarization shifts.
The implementation of single-cell omics technologies (e.g., transcriptomics, proteomics, and metabolomics) could generate significant insights into the molecular pathways that drive macrophage polarization in LN, as well as identify new pathways and molecular signatures that may serve as therapeutic targets. However, translating macrophage-targeted therapies from preclinical studies to clinical practice in LN settings present significant challenges due to differences between animal models and human disease, dynamic macrophage behavior, and patient heterogeneity. Biomarkers for monitoring macrophage activity in patients remain underdeveloped, and immunomodulators can have off-target effects. In addition, the complexity of LN suggests a need for multifaceted approaches that provide opportunities to modulate macrophage polarization while at the same time targeting the inhibition of pro-inflammatory cytokines or enhancing anti-inflammatory responses. Future research should focus on humanized models, combination therapies, advanced imaging, and personalized approaches to improve clinical translation. Characterization of these biomarkers in preclinical and clinical studies are needed to validate our observations, and based on these findings, establish protocols to evaluate the appropriate safety and efficacy of macrophage-targeted therapy for patients with LN, and establish best-practice assessment protocols in the clinic for conducting standard measurements. The goal for the treatment of LN is personalized medicine, which aims to tailor therapies based on individual patient factors like macrophage polarization, cytokine profiles, and genetic predispositions to attain the best possible outcomes with minimal side effects/discontinuation of therapy.
This review focuses on the key aspects of macrophage polarization in LN pathogenesis and management, highlighting the opposing roles of M1 macrophages, which promote inflammation, and M2 macrophages, which promote tissue repair and anti-inflammatory responses. Macrophage functions and LN are modulated through a dynamic process involving numerous microenvironment components. Furthermore, new strategies implicated in ameliorating renal inflammation and improving clinical outcomes target macrophage polarization and signaling pathways. The identification of biomarkers to facilitate early diagnosis in conjunction with therapies that alter macrophage function, such as M2 macrophage transfusions, represent promising approaches for the more effective treatment of LN. While current treatments for LN have variable efficacy, furthering our understanding of the context-dependent contributions of macrophage function is critical to advancing therapeutics that will reduce inflammation but also improve tissue repair and restore immune homeostasis.
LZ: Writing-original draft and Visualization. JS: Substantial contributions to the conception and supervision; TL: Writing-review & editing and Conceptualization. LZ conceived the study, designed the methodology. JS participated in manuscript review and editing. TL oversaw project administration. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.