
- Academic Editors
Glutamate excitotoxicity is one of the key factors in the pathophysiology of the secondary injury cascade following traumatic brain injury (TBI) and spinal cord injury (SCI). These neurotraumatic conditions remain major causes of long-term disability and mortality worldwide, yet therapeutic options remain limited. Excessive glutamate release after neurotrauma leads to the overactivation of glutamate receptors, triggering calcium influx and the activation of destructive enzymes and signaling pathways that drive progressive neuronal death and tissue degeneration. This review examines the molecular mechanisms of glutamate-mediated excitotoxicity in neurotrauma, particularly focusing on TBI and SCI, and evaluates current and emerging therapeutic strategies aimed at modulating glutamate levels, receptor activity, and downstream signaling pathways. Particular attention is given to glutamate receptor antagonists, agents enhancing glutamate clearance, and neuroprotective compounds. A critical analysis of preclinical successes versus clinical failures reveals key translational barriers, including narrow therapeutic windows, patient heterogeneity, poor drug penetration across the blood-brain barrier, and adverse off-target effects. Delayed treatment relative to the peak of excitotoxic activity has also limited clinical efficacy. This review highlights the importance of understanding the temporal dynamics of glutamate toxicity and the necessity for precisely timed, stratified therapeutic interventions. This work contributes to the broader scientific effort to develop more effective neuroprotective therapies by identifying the mechanistic underpinnings and translational challenges of anti-excitotoxic strategies. Given the global burden of TBI and SCI, advancing targeted interventions for glutamate excitotoxicity holds significant promise for improving neurological outcomes and quality of life for affected individuals.
Neurotrauma, encompassing traumatic brain injury (TBI) and spinal cord injury (SCI), represents a significant global health challenge, ranking among the leading causes of morbidity and mortality worldwide [1, 2, 3, 4, 5, 6, 7]. These injuries contribute to substantial economic and social burdens, including long-term disability, loss of productivity, and considerable healthcare costs [8]. Globally, the incidence of TBI and SCI is particularly high in low- and middle-income countries, where limited access to emergency care and rehabilitation services exacerbates the outcomes [9, 10]. The heterogeneity in injury severity, mechanisms, and individual responses to treatment further complicates clinical management.
The pathophysiology of neurotrauma is multifaceted, involving an immediate primary injury caused by mechanical forces and a subsequent secondary injury cascade that unfolds over hours to weeks post-trauma [11, 12]. The primary injury results from the physical disruption of tissues, including neuronal and vascular components. In contrast, the secondary injury cascade encompasses ischemia, oxidative stress, inflammation, edema, and excitotoxicity, which propagate cellular damage, amplify neurodegeneration, and hinder reparative processes [13, 14, 15, 16, 17].
Among these mechanisms, glutamate excitotoxicity has emerged as a critical contributor to secondary injury. Following neurotrauma, the excessive release of glutamate, a major excitatory neurotransmitter, overstimulates ionotropic glutamate receptors such as N-methyl-D-aspartate (NMDA) and alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors [18, 19, 20]. This overactivation leads to a pathological influx of calcium ions into neurons, triggering enzymatic cascades that degrade cellular structures, including membranes and cytoskeletal proteins [19, 20]. Concurrently, calcium overload in mitochondria impairs energy production, exacerbates Reactive Oxygen Species (ROS) generation, and initiates apoptotic and necrotic pathways [21].
The deleterious effects of excitotoxicity extend beyond neuronal death, contributing to white matter degeneration, disruption of axonal integrity, and the formation of cystic cavities, which impede functional recovery [16, 22, 23, 24]. ROS further amplifies these processes by damaging lipids, proteins, and nucleic acids, overwhelming the antioxidant defense mechanisms of injured tissues [19].
Addressing glutamate excitotoxicity is therefore paramount in the development of neuroprotective interventions for neurotrauma, particularly in TBI and SCI. This review consolidates recent advances in understanding the mechanisms of glutamate-induced neurotoxicity and its central role in exacerbating secondary injury following central nervous system (CNS) trauma. Specifically, it explores therapeutic strategies aimed at mitigating excitotoxicity, including glutamate receptor antagonists, agents that enhance glutamate clearance, and compounds that target downstream pathways implicated in neuronal damage.
Despite encouraging results in preclinical studies, the clinical translation of anti-excitotoxic therapies remains limited. Several factors contribute to this persistent gap, including species-specific responses, the use of animal models that inadequately reflect human injury heterogeneity and comorbidities, and the delayed timing of therapeutic administration in clinical settings. Notably, the transient and dynamic nature of excitotoxic cascades underscores the importance of therapeutic timing—many interventions, such as NMDA receptor antagonists, are only effective within a narrow post-injury window. This highlights the need for precision-targeted strategies that account for the temporal evolution of excitotoxicity.
Additionally, this review emphasizes the broader clinical and global relevance of glutamate-targeting interventions. Given the substantial burden of neurotrauma, particularly in underserved regions, there is an urgent need for scalable and effective treatments. By integrating mechanistic insights with emerging therapeutic evidence, this review aims to guide the development of targeted interventions—pharmacological, antioxidant, and neuroprotective—that can be more effectively translated into clinical practice and ultimately improve outcomes for individuals with TBI and SCI.
Glutamate serves as the principal excitatory neurotransmitter in the mammalian CNS [25, 26, 27, 28, 29, 30], playing a vital role in synaptic transmission (Figs. 1,2). Stored in synaptic vesicles, glutamate is released into the synaptic cleft primarily through exocytosis, a process triggered by calcium influx at nerve terminals during neuronal activation [30] (Fig. 2). Beyond this primary mechanism, alternative pathways, such as anion channels and reverse operation of glutamate transporters, also contribute to extracellular glutamate levels. Once released, glutamate activates two classes of receptors: ionotropic and metabotropic (Fig. 1). Ionotropic receptors, including NMDA, AMPA, and kainate receptors, mediate rapid synaptic responses and calcium influx, with NMDA receptors being key contributors to excitotoxicity. Metabotropic receptors, which operate through G-protein signaling, modulate slower, intracellular pathways and play diverse roles in synaptic regulation. These receptors activate a range of downstream signaling cascades, such as calcium/calmodulin-dependent protein kinase II (CaMKII), phospholipase C, mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK), and phosphatidylinositol-3 kinase/protein kinase B (PI3K/AKT) pathways, which govern processes like synaptic plasticity, gene expression, and cell survival.
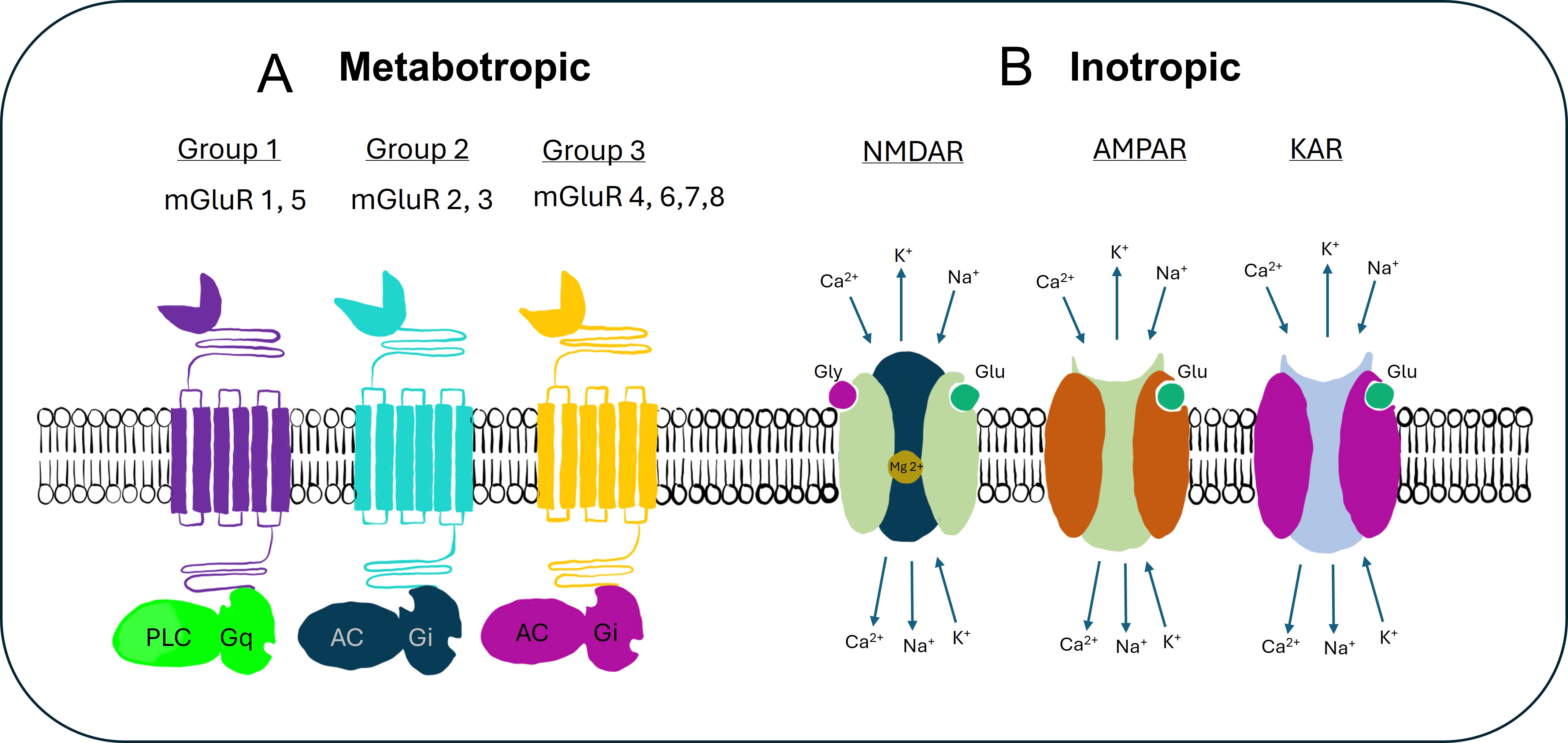 Fig. 1.
Fig. 1.
Glutamate receptors. This figure illustrates the key types of
glutamate receptors involved in neurotransmission. (A) Highlights the
metabotropic glutamate receptors (mGluRs), which are G-protein-coupled receptors
that modulate neuronal excitability and synaptic transmission over a slower time
scale. mGluRs are classified into three groups (1, 2, and 3) based on their
signaling pathways and effects on the postsynaptic cell. The roles of these
receptors are in synaptic plasticity, learning, and neurodevelopment. (B) Shows
the ionotropic glutamate receptors, including NMDA (N-methyl-D-aspartate)
receptors, AMPA
(
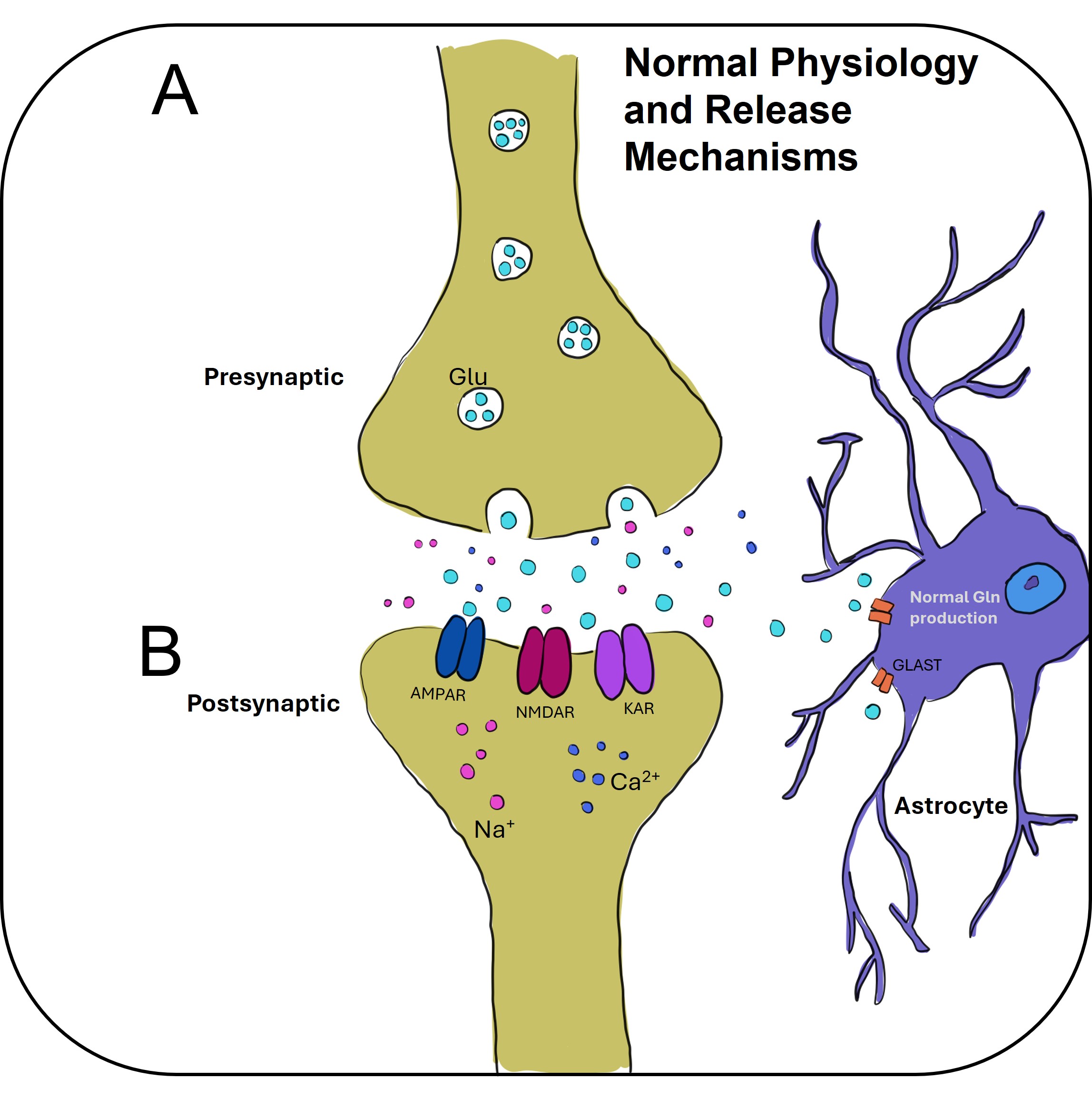 Fig. 2.
Fig. 2.
This figure depicts the physiological processes involved in glutamate release and its subsequent action at the synapse. (A) Illustrates the process of glutamate release from the presynaptic neuron. Glutamate, stored in vesicles within the presynaptic terminal, is released into the synaptic cleft in response to an action potential. The release is mediated by voltage-gated calcium channels, which trigger vesicle fusion with the presynaptic membrane, allowing glutamate to flow into the synapse. (B) Shows the action of glutamate on the postsynaptic receptors, including ionotropic receptors (e.g., NMDA, AMPA, and Kainate receptors) and metabotropic glutamate receptors (mGluRs). Upon binding, glutamate activates these receptors, leading to either fast excitatory postsynaptic potentials (via ionotropic receptors) or slower modulatory effects (via mGluRs). The regulation of glutamate release and signaling plays a crucial role in synaptic plasticity, learning, and neurodevelopment, as well as in diseases related to excitatory neurotransmission. Created with Adobe Fresco.
The role of glutamate in synaptic plasticity is particularly significant in
phenomena like long-term potentiation (LTP) and long-term depression (LTD), which
are crucial for learning and memory. However, excessive glutamate activity can
result in excitotoxicity, leading to neuronal damage and neurodegenerative
conditions (Fig. 3). To maintain homeostasis, glutamate clearance from the
synaptic cleft is managed by excitatory amino acid transporters (EAATs),
predominantly EAAT1 (GLAST) and EAAT2 (GLT-1), which are highly expressed in
astrocytes. Once cleared, glutamate is either recycled as glutamine for reuse in
neurotransmission or metabolized into
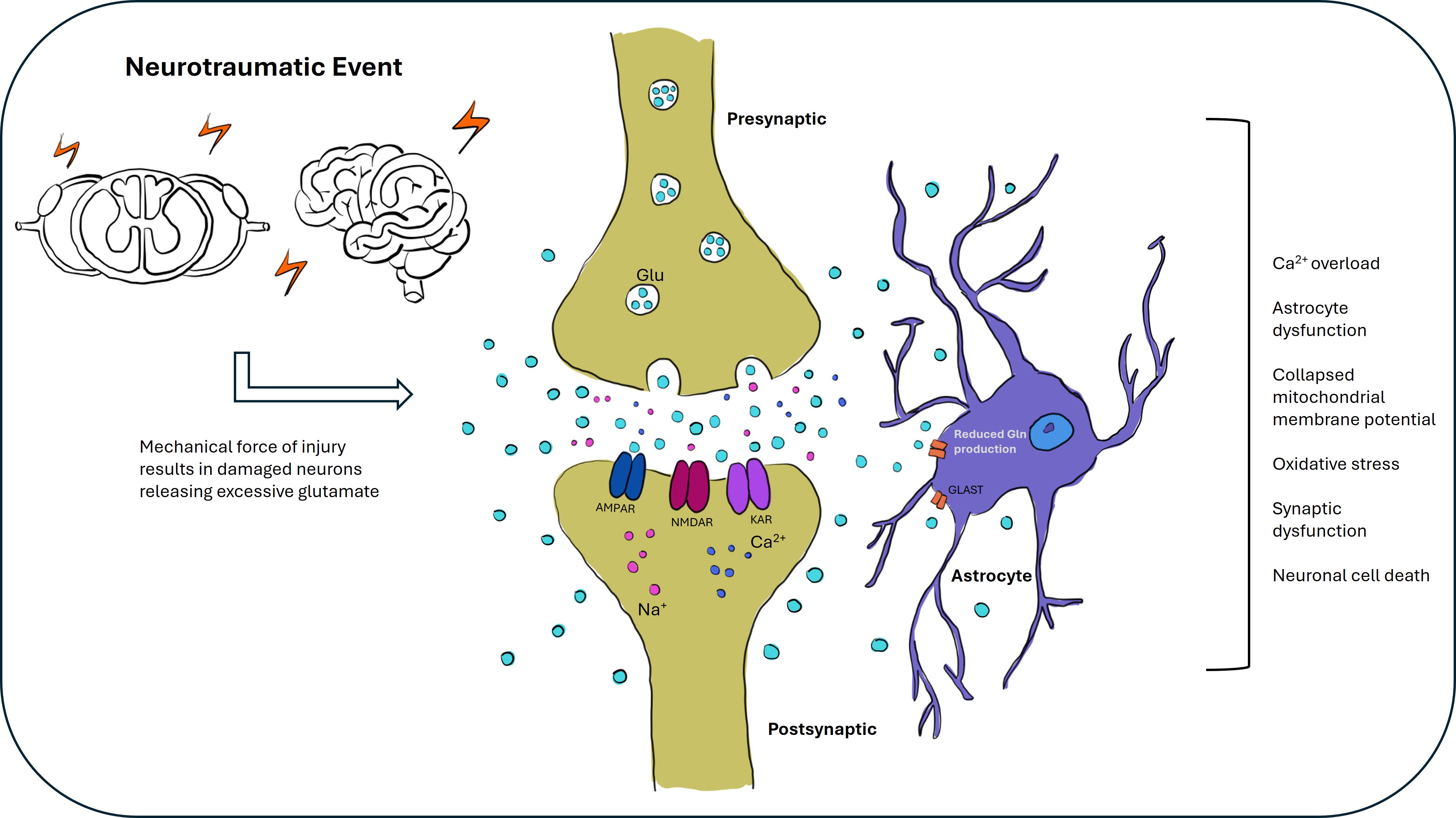 Fig. 3.
Fig. 3.
Glutamate excitotoxicity in brain and spinal cord injury. This figure illustrates the role of glutamate excitotoxicity following brain and spinal cord injury (SCI). Following an injury to the brain or spinal cord, the blood-brain barrier (BBB) is often compromised, and there is an increased release of glutamate into the extracellular space due to neuroinflammation and glial cell activation. Excessive glutamate activates NMDA receptors, causing calcium influx, which triggers a cascade of harmful intracellular events, including oxidative stress, mitochondrial dysfunction, and cell death. This process contributes to neuronal damage and worsens secondary injury in both the brain and spinal cord. In response to injury, glutamate transporters can become overwhelmed or dysfunctional, exacerbating excitotoxicity. The figure highlights the critical role of glutamate excitotoxicity in the pathophysiology of brain and spinal cord injury and its potential as a target for therapeutic intervention to prevent further neuronal damage and promote recovery. Created with Adobe Fresco.
Glutamate excitotoxicity refers to the pathological process whereby excessive or sustained activation of glutamate receptors leads to toxic cellular outcomes. This phenomenon is characterized by an overwhelming influx of ions, particularly calcium, into neurons, which activates a cascade of deleterious enzymatic processes. Key enzymes, such as proteases, phospholipases, and endonucleases, are triggered, resulting in the degradation of cellular structures, disruption of normal neuronal function, and eventual cell death [27, 31]. This mechanism underlies many neurodegenerative and neurotraumatic conditions (Fig. 3), highlighting the critical need to regulate glutamate signaling to prevent excitotoxic damage.
The concept of glutamate excitotoxicity has its roots in the mid-20th century. In 1954, Hayashi [32] first reported the seizure-inducing effects of glutamate by applying high concentrations of sodium glutamate directly, providing early evidence of its excitatory and potentially harmful properties. Subsequently, in 1957, Lucas and Newhouse [33] demonstrated the toxic effects of glutamate in a different context by showing degeneration of the retina’s inner layers following glutamate injections in infant mice. This pivotal finding highlighted glutamate’s neurotoxic potential in specific concentrations and locations. The term “glutamate excitotoxicity” was later introduced by Olney in 1969 [34], formally identifying the pathological process and establishing its significance in neuroscience.
Neurotrauma, including TBI and SCI, remains a significant cause of neurological disability worldwide [13]. Beyond the primary mechanical damage, secondary injury processes such as ischemia, oxidative stress, inflammation, and glutamate excitotoxicity exacerbate cellular damage and hinder the effectiveness of therapeutic interventions aimed at neural repair [13, 14, 15, 16, 17]. This discussion centers on elucidating the mechanisms underlying glutamate excitotoxicity in the context of neurotrauma, emphasizing its pivotal role in secondary injury (Fig. 3) and its implications for developing targeted treatments.
Growing evidence highlights the influence of sex on the pathophysiological
response to TBI and SCI, with implications for both injury progression and
therapeutic outcomes. Preclinical and clinical studies increasingly demonstrate
that sex differences affect injury severity, recovery trajectories, and treatment
efficacy. One notable clinical study by Wagner et al. [35] examined the
interplay between sex, age, and therapeutic hypothermia on markers of
excitotoxicity, oxidative stress, and ischemia following severe TBI.
Cerebrospinal fluid (CSF) samples from 68 adults (Glasgow Coma Scale
Complementary studies employing microdialysis and magnetic resonance spectroscopy have reinforced the clinical significance of glutamate as both a biomarker and a driver of injury. Elevated extracellular glutamate levels have been consistently observed in the acute phase post-injury, with concentrations correlating with injury severity and long-term neurological outcomes. Clinical studies by Chamoun et al. [36] and Bullock et al. [37] further demonstrated that persistently high glutamate levels are associated with increased mortality and poorer recovery, validating glutamate as a relevant therapeutic target. Together, these data reinforce the translational importance of targeting glutamate excitotoxicity and advocate for individualized treatment approaches that account for sex, age, and injury-specific dynamics.
Traumatic injury directly impacts neuronal integrity, causing axonal shearing, neuronal rupture, and bleeding, which elevate extracellular concentrations of excitatory amino acids such as glutamate [13, 38, 39, 40, 41]. Under normal conditions, extracellular glutamate concentration is approximately 0.6 µmol/L [41]. Following trauma, this level can rise tenfold to around 10 µmol/L due to the release of intracellular glutamate from damaged neurons [41]. This surge in glutamate concentration places surrounding neurons at risk, triggering a cascade of excitotoxic events that propagate neuronal death and amplify tissue damage. NMDA-receptor antagonists have demonstrated neuroprotection in experimental neurotrauma models, supporting the hypothesis that excitatory amino acid efflux significantly contributes to neural damage following TBI [39].
The unique shearing forces associated with TBI, directly and indirectly, cause secondary injury through glutamate excitotoxicity (Fig. 3). Excess glutamate overstimulates glutamate receptors, particularly ionotropic NMDA receptors, leading to a massive influx of calcium ions (Ca2+) into neurons [5]. Notably, NMDA receptors are mechanosensitive, a characteristic distinct to this injury mechanism [42]. Kainic acid (KA) and AMPA receptors, which are partially permeable to calcium, further contribute to intracellular calcium overload [43]. This unregulated Ca2+ accumulation activates destructive enzymes, including proteases, lipases, and endonucleases, and promotes the generation of ROS, exacerbating cellular damage and leading to cell death [35, 44, 45, 46, 47]. Additionally, calcium influx depolarizes the neuronal membrane, triggering voltage-dependent calcium channels (e.g., N-type or P/Q-type) to further amplify intracellular calcium levels [48, 49].
Glutamate synthesis occurs through the Krebs cycle in mitochondria or via the glutamine-glutamate cycle [30]. After synthesis, glutamate is stored in synaptic vesicles and released during exocytosis. Calcium-sensing proteins such as synaptotagmin-1 detect elevated intracellular Ca2+ and activate the SNARE complex, facilitating vesicle fusion with the cell membrane and releasing more glutamate [30]. This abnormal release triggers a pathological feedback loop, causing further glutamate release and perpetuating excitotoxicity [41].
The clearance of glutamate is a critical process for maintaining proper
glutamatergic signaling and preventing excitotoxicity. In the CNS, glutamate is
primarily contained within cells, as there are no enzymes capable of metabolizing
glutamate in the extracellular space. The removal of glutamate from the synaptic
cleft relies on EAATs, predominantly expressed in astrocytes and nearby glial
cells [50, 51, 52] (Figs. 2,3). The main astrocytic glutamate transporters, EAAT1
(GLAST) and EAAT2 (GLT-1), play pivotal roles in this process, with EAAT2 being
responsible for the majority of glutamate uptake across brain regions [50, 53].
Once transported into astrocytes or neurons, glutamate is either converted to
Following neurotrauma, however, intracellular signaling alterations impair the localization and function of glutamate transporters. Studies indicate that glutamate transporters, particularly EAAT2, are downregulated within 24 hours post- TBI [5, 56]. This reduction in transporter availability and efficiency leads to impaired glutamate reuptake, resulting in elevated extracellular glutamate levels and the dysregulation of synaptic homeostasis. Consequently, the persistence of elevated glutamate in the extracellular space exacerbates excitotoxicity, further damaging neurons and surrounding neural tissue.
In the face of such dysregulation, both astrocytes and neurons deploy compensatory mechanisms aimed at counteracting excessive extracellular glutamate and limiting excitotoxic damage. Astrocytes, as the primary mediators of glutamate clearance, can upregulate other glutamate-buffering proteins under pathological stress. One key player is glutamine synthetase (GS), an astrocyte-specific enzyme that catalyzes the conversion of glutamate to glutamine. This enzymatic pathway provides a critical alternative route for detoxifying surplus glutamate and has been shown to be upregulated in response to elevated glutamate levels [57, 58, 59]. In parallel, astrocytes can undergo morphological adaptations—such as hypertrophy and elongation of processes—that expand their glutamate uptake capacity and enhance spatial buffering [60].
Furthermore, under certain pathological conditions, reactive astrocytes may transiently alter their EAAT isoform expression. For example, despite the general downregulation of EAAT2 following injury, some studies have reported context-dependent upregulation of this transporter as part of a neuroprotective response [61, 62]. These plastic changes in astrocytic behavior represent a dynamic attempt to restore glutamate homeostasis and limit neurotoxicity.
On the neuronal side, homeostatic responses also play a crucial role in buffering excitotoxic stress. Neurons exposed to prolonged glutamate stimulation may downregulate postsynaptic receptor expression or modify receptor subunit composition to reduce calcium influx. For instance, sustained activation of NMDA receptors can trigger signaling pathways that internalize receptors or favor subunit configurations with lower calcium permeability [63]. Similarly, AMPA receptors can incorporate GluA2 subunits under chronic stimulation, rendering them calcium-impermeable and more resistant to excitotoxic injury [64, 65].
These adaptive mechanisms—spanning molecular, structural, and synaptic levels—reflect the CNS’s intrinsic ability to restore balance in the face of excitatory dysregulation. Evidence from models of chronic neurodegeneration and ischemia further suggests that such compensatory responses play a pivotal role in neuronal survival under sustained excitotoxic threat [66, 67]. Understanding these endogenous protective strategies provides valuable insights into therapeutic targets that could bolster the CNS’s resilience following injury and reduce the burden of neurotrauma-induced damage.
The excessive influx of Ca2+ through glutamate receptors following neurotrauma leads to abnormal calcium accumulation within mitochondria. This disrupts mitochondrial function by uncoupling the electron transport chain from ATP synthesis, impairing energy metabolism, and significantly increasing the production of free radicals [68, 69, 70]. These reactive oxygen species exacerbate oxidative stress and contribute to further neuronal damage.
Additionally, mitochondrial dysfunction and the consequent ATP depletion impair the activity of GS, a crucial ATP-dependent enzyme. GS converts glutamate to glutamine, maintaining glutamate homeostasis and preventing excitotoxicity. Following injury, decreased ATP levels limit the functionality of GS, further disrupting glutamate clearance and amplifying excitotoxic damage [71].
Glutamate excitotoxicity is intricately linked to neuroinflammation, with a bidirectional and self-reinforcing relationship between the two processes. Following neurotrauma, the neuronal damage induced by excessive glutamate release triggers the release of damage-associated molecular patterns (DAMPs), which activate microglia and astrocytes [72, 73]. These activated glial cells release a variety of pro-inflammatory cytokines, chemokines, and other inflammatory mediators [72, 73]. In turn, these inflammatory mediators contribute to the further release of glutamate and the inhibition of glutamate uptake by astrocytes, thereby exacerbating the already elevated extracellular glutamate levels. This cycle of increased glutamate release and impaired glutamate clearance fuels both excitotoxicity and neuroinflammation, creating a vicious cycle that amplifies neuronal damage and sustains secondary injury processes.
Following CNS injury, this interplay between excitotoxicity and
neuroinflammation becomes a central driver of secondary neuronal damage. Excess
extracellular glutamate overstimulates ionotropic receptors, particularly NMDA
and AMPA receptors, resulting in excessive calcium influx, mitochondrial
dysfunction, and neuronal death [74]. This excitotoxic environment rapidly
activates resident glial cells—microglia and astrocytes—which further
propagate neuroinflammatory responses. Microglia, the brain’s primary immune
cells, rapidly transition from a surveillant to an activated state post-injury.
Activated microglia release pro-inflammatory cytokines such as TNF-
Prolonged glial activation also promotes glial scar formation and chronic inflammation, both of which hinder regeneration and repair. Nonetheless, glial responses are not uniformly detrimental. Under specific conditions, microglia and astrocytes can adopt neuroprotective phenotypes, secreting anti-inflammatory mediators and supporting tissue remodeling [77]. Thus, the dual role of glial cells highlights the need for therapeutic strategies that modulate, rather than completely suppress, glial activation—aiming to mitigate excitotoxic damage while facilitating central nervous system repair.
Current therapeutic strategies for managing glutamate excitotoxicity following TBI or SCI aim to reduce excessive glutamate release, block its receptors, and protect neurons from the downstream effects of excitotoxicity. Several potential therapies have been explored, targeting different stages of the glutamate excitotoxicity cascade (detailed in Table 1 (Ref. [78, 79, 80, 81, 82, 83, 84, 85] below).
| Agent | Mechanism of action | Preclinical findings | Clinical findings | Limitations |
| Dizocilpine (MK-801) | Non-competitive NMDA receptor antagonist | Demonstrated neuroprotection in animal models of ischemia and spinal cord injury; improved neurological recovery when administered post-injury [78]. | Clinical trials halted due to severe side effects, including hallucinations and neurotoxicity (Olney’s lesions) [79]. | High-affinity NMDA blockade interferes with normal synaptic function; significant psychotomimetic effects limit clinical utility. |
| Memantine | Low-affinity, uncompetitive NMDA receptor antagonist; preferentially blocks extrasynaptic receptors | Exhibited neuroprotective effects in animal models by reducing calcium influx and excitotoxicity; preserves normal synaptic transmission [80]. | Approved for Alzheimer’s disease; limited evidence for efficacy in acute CNS injuries like TBI or SCI [81]. | Benefits in acute neurotrauma remain unproven; primarily used in chronic neurodegenerative conditions. |
| Nitromemantine | Memantine derivative targeting extrasynaptic NMDA receptors; includes nitric oxide moiety | Enhanced neuroprotection and synaptic preservation in preclinical Alzheimer’s models; reduced excitotoxicity with fewer side effects [82, 83]. | Not yet evaluated in clinical trials. | Requires further research to establish safety and efficacy in humans. |
| Riluzole | Inhibits glutamate release; enhances glutamate uptake; modulates sodium channels | Reduced neuronal damage in animal models of ALS and spinal cord injury; decreased glutamate-induced excitotoxicity [84]. | Approved for ALS; clinical trials in TBI and SCI have shown mixed results, with some improvements in neurological outcomes [84]. | Modest clinical benefits; side effects include liver dysfunction and fatigue. |
| GCPII Inhibitors (e.g., 2-PMPA) | Inhibit glutamate carboxypeptidase II to prevent NAAG breakdown, reducing glutamate levels | Provided neuroprotection in animal models of stroke, TBI, and neuropathic pain by lowering extracellular glutamate [85]. | No clinical trials conducted to date. | Poor blood-brain barrier penetration; pharmacokinetic challenges hinder clinical development. |
CNS, central nervous system; TBI, traumatic brain injury; SCI, spinal cord injury; ALS, Amyotrophic Lateral Sclerosis; GCPII, Glutamate Carboxypeptidase II; 2-PMPA, 2-(Phosphonomethyl) pentanedioic acid; NAAG, N-Acetylaspartylglutamate.
This table highlights the complexity of translating preclinical successes into effective clinical therapies for glutamate excitotoxicity. While several agents show promise in animal models, their clinical applicability is often limited by side effects, pharmacokinetic issues, or lack of efficacy in human trials. Several ongoing research studies aim to develop safer, more targeted treatments that can effectively mitigate excitotoxic damage in CNS injuries.
Compounds that selectively block NMDA receptors can help prevent excessive
calcium influx—a key event in glutamate-induced excitotoxicity. NMDA receptor
antagonists like amantadine and memantine have demonstrated positive effects in
clinical trials with TBI patients [5, 86]. The beneficial effects of ketamine, an
NMDA receptor antagonist, in experimental head injury have been demonstrated,
emphasizing the potential of NMDA receptor modulation in TBI treatment [87].
Selective NR2B subunit antagonists, such as ifenprodil and taxoprodil, have shown
improvements in mortality and injury volume in TBI patients and animal models
[88, 89, 90]. However, the potential for cognitive impairment warrants caution in
their use. AMPA/kainate receptor blockers, such as perampanel, have also shown
acute neuroprotective effects after TBI [91, 92]. Additionally, the protective
effect of memantine, a well-established NMDA receptor antagonist, against
A
Agents that inhibit glutamate release from presynaptic terminals can reduce the overall glutamate burden in the extracellular space. This may involve targeting voltage-gated calcium channels or vesicular glutamate transporters. Several studies have investigated various compounds and their effects on glutamate release. For instance, allopregnanolone has been shown to inhibit glutamate release from cerebrocortical nerve terminals, and this effect was abolished by inhibitors of Ca2+/calmodulin, adenylate cyclase, and protein kinase A [94]. Similarly, typhaneoside was found to suppress glutamate release through the inhibition of voltage-dependent calcium entry in rat cerebrocortical nerve terminals [95]. Furthermore, cycloheterophyllin and lycopene were reported to inhibit glutamate release through the suppression of voltage-dependent Ca2+ entry and protein kinase C, demonstrating their potential as glutamate release inhibitors [96, 97]. Moreover, Riluzole, a glutamate-release inhibitor, has exhibited neuroprotection following SCI and TBI by blocking voltage-dependent sodium channels [98, 99]. Furthermore, Ceftriaxone, a beta-lactam antibiotic, enhances glutamate clearance by upregulating glutamate transporters (EAAT2) and has shown promise in reducing brain trauma in animal studies [100, 101].
Excessive calcium influx is a key contributor to excitotoxicity. Calcium channel blockers like ziconotide, a Voltage-Gated Calcium Channel (VGCC) antagonist, have been shown to reduce Ca2+ accumulation and improve motor and cognitive function in preclinical models of TBI [102, 103], although cardiovascular side effects halted clinical trials [104]. Nimodipine, an L-type calcium channel blocker, has also demonstrated neuroprotective effects in models of cerebral ischemia [105]. Ryanodine receptors (RyRs), which release Ca2+ from intracellular stores, are implicated in excitotoxicity. Dantrolene, a RyR antagonist, has shown protective effects in TBI models by inhibiting abnormal calcium release [106, 107].
Since neuroinflammation often accompanies TBI, anti-inflammatory agents may indirectly reduce excitotoxicity. Inflammation can enhance glutamate release and decrease glutamate uptake, exacerbating excitotoxicity. N-acetylcysteine, flavonoids, and phenelzine have shown therapeutic effects by acting as antioxidants and scavenging trauma-induced free radicals [108, 109, 110, 111]. Taurine, the most abundant free amino acid in the brain, can induce cellular hyperpolarization through GABAA and glycine receptors, reducing NMDA-mediated excitotoxicity and calcium overload [112]. It also mitigates mitochondrial stress and apoptosis. Minocycline, a tetracycline antibiotic, has been shown to reduce neuroinflammation and oxidative stress, though clinical trials have yielded mixed results [113, 114, 115, 116]. Synthetic peroxisome proliferator-activated receptor (PPAR) agonists have been identified as potent anti-inflammatory therapeutic agents for TBI [117]. Additionally, recent literature suggests that drugs such as minocycline and phenytoin may serve as worthwhile repositioned therapeutics in treating TBI, indicating their potential in addressing inflammation and excitotoxicity [118]. Furthermore, anti-inflammatory agents such as glutathione peroxidase have been identified as potential therapeutic targets for pediatric TBI, emphasizing the role of anti-inflammatory strategies in TBI management [119]. Cox inhibitor. Several articles highlight the potential role of selective cyclooxygenase-2 (COX-2) inhibitors in mitigating glutamate-induced neurotoxicity following CNS injuries [120, 121, 122]. After CNS injury, COX-2 expression is upregulated, leading to increased production of pro-inflammatory prostaglandins that contribute to neuronal damage. Selective COX-2 inhibitors, such as celecoxib and nimesulide, may attenuate glutamate-mediated excitotoxicity and reduce neuroinflammation, thereby offering neuroprotection. The articles conclude that selective COX-2 inhibitors hold potential as therapeutic agents to mitigate glutamate-induced neurotoxicity following CNS injuries. While preclinical studies are promising, further clinical trials are essential to confirm the efficacy and safety of COX-2 inhibitors in human CNS injury contexts.
Neurotrophic factors and mitochondrial protectants, such as coenzyme Q10 (CoQ10), have demonstrated counteracting effects on excitotoxic damage [123, 124]. The sigma-1 receptor has been identified as a potential therapeutic target for TBI due to its wide distribution in the body and its ability to interact with various drug structural classes [125]. Sigma-1 receptor agonists have shown promise as a therapeutic strategy for brain injury, with multiple mechanisms of action, including the stabilization of mitochondrial membrane potential, inhibition of microglial activation, and reduction of cell death [126]. Activation of sigma-1 receptors has been found to promote neuroprotection after ischemic and traumatic injuries to the central nervous system [127]. Another approach involves targeting mitochondrial dysfunction using agents such as Methylene Blue, which has shown promise in ameliorating mitochondrial dysfunction following brain injury [128]. Additionally, cyclosporine, a mitochondrial permeability transition pore inhibitor, has demonstrated benefits in improving mitochondrial function and reducing tissue damage in experimental TBI models [129]. Another therapeutic, pioglitazone has been shown to restore mitochondrial homeostasis and promote functional recovery after TBI, indicating its potential as a therapeutic agent [130]. Moreover, targeting specific proteins such as SIRT3 and PRDX3 has been identified as a significant downstream strategy to reduce oxidative damage following TBI, highlighting the potential of these proteins as therapeutic targets [131].
Although many of these agents show promise in preclinical models of neurotrauma and excitotoxicity-induced neurological disorders, clinical trial results remain mixed, and side effects are a concern. Future research should focus on identifying the most effective therapies with minimal side effects. Combining different approaches targeting various stages of excitotoxicity may offer more effective treatments. Additionally, the timing of therapeutic initiation is crucial, as many treatments are more effective when administered early in the excitotoxicity cascade. Immediate treatment with glutamate receptor blockers, for instance, can reduce the spread of neuronal death after injury [41]. The underlying cause of excitotoxicity, such as SCI or stroke, may also influence the choice and efficacy of specific treatments.
In the immediate phase (minutes to hours post-injury) (Fig. 4), excessive glutamate release rapidly overactivates NMDA receptors, causing calcium overload and neuronal injury. NMDA receptor antagonists show the greatest neuroprotective efficacy when administered within the first few hours after TBI, effectively attenuating the excitotoxic cascade [132]. During the delayed phase (24–72 hours post-injury), NMDA receptor expression declines, and partial NMDA receptor agonists such as D-cycloserine have demonstrated neuroprotective and functional benefits when administered within this subacute window [132]. In the acute phase (within 15 minutes post-injury), a surge in extracellular glutamate similarly drives excitotoxic loss of neurons and oligodendrocytes. Early administration of AMPA receptor antagonists, such as topiramate, within minutes after injury has been shown to promote tissue preservation and improve motor outcomes [133]. Together, these findings highlight the importance of timing and receptor-specific targeting in mitigating excitotoxicity and inflammation across both acute and secondary phases of CNS trauma (as illustrated in Fig. 4). Extended Window (Up to 4 Hours Post-Injury): Although earlier intervention yields better outcomes, some efficacy is still observed with delayed administration of AMPA/kainate receptor antagonists (e.g., NBQX) up to four hours post-injury, offering a slightly broader therapeutic window [133].
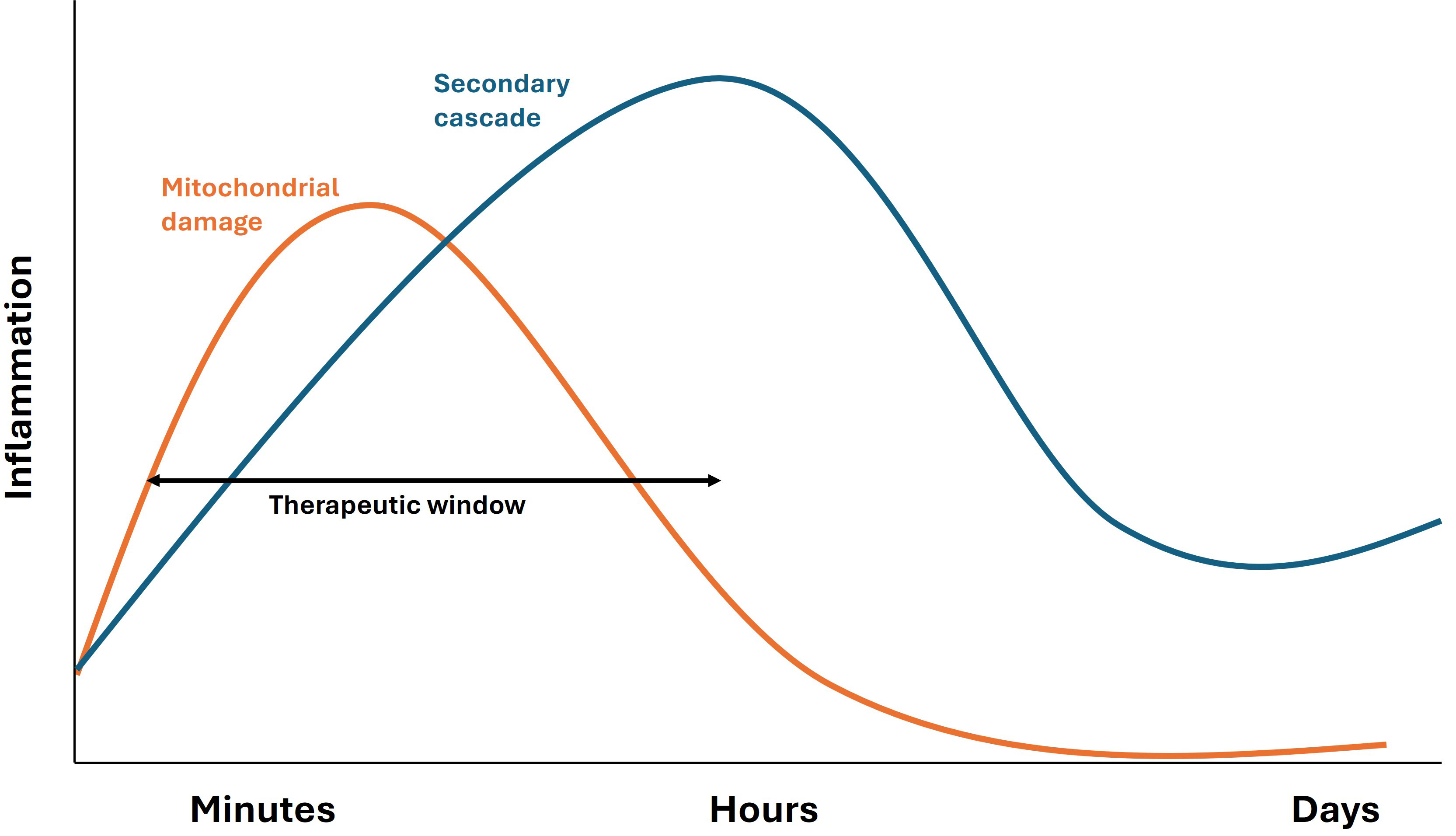 Fig. 4.
Fig. 4.
Temporal profile of mitochondrial damage and secondary inflammatory cascades following TBI and SCI. This schematic illustrates the biphasic pattern of inflammation after TBI or SCI. The initial phase (orange curve) represents acute mitochondrial damage, which triggers oxidative stress, energy failure, and an early surge of pro-inflammatory mediators. This phase contributes to neuronal and glial dysfunction immediately after injury. As the acute response diminishes, a secondary inflammatory cascade (blue curve) develops, characterized by activation of microglia, astrocytes, and infiltrating immune cells. This secondary wave sustains chronic neuroinflammation, promotes secondary neuronal degeneration, and contributes to long-term functional deficits. Understanding these temporally distinct but overlapping inflammatory phases is critical for developing therapeutic interventions targeting both acute and chronic stages of CNS injury. Preclinical studies consistently highlight the importance of early intervention to mitigate glutamate excitotoxicity and secondary injury following TBI and SCI. However, translating these findings into clinical practice remains challenging. The rapid evolution of secondary injury mechanisms post-injury underscores the urgency for timely therapeutic strategies.
The concept of a therapeutic window in neurotrauma centers around the evolving secondary injury cascade initiated by the primary mechanical insult. While the primary injury occurs within seconds and is largely irreversible, the secondary processes—which unfold over hours to days—present a critical opportunity for therapeutic intervention [134, 135].
Early Excitotoxicity (Within 1–3 Hours): The initial insult rapidly increases intracellular calcium via glutamate receptor overactivation, triggering activation of proteases, lipases, and endonucleases, and leading to ROS generation [136, 137]. These pathological events peak within the first few hours post-injury, suggesting that therapies targeting voltage-gated calcium channels or vesicular glutamate transporters would be most effective during this acute window [138].
Mitochondrial Dysfunction (30 Minutes to 6 Hours): Mitochondria become compromised due to calcium overload and oxidative stress, resulting in the opening of the mitochondrial permeability transition pore, membrane depolarization, and cytochrome c release—activating intrinsic apoptotic pathways [139, 140]. Mitochondrial dysfunction becomes evident within 30 minutes and progressively worsens, making this period optimal for administering mitochondrial stabilizers, antioxidants, anti-inflammatory agents, and other neuroprotective compounds.
Inflammatory Response (6–72 Hours): A delayed yet sustained inflammatory response begins to dominate, marked by microglial activation, cytokine release, and leukocyte infiltration across a compromised blood-brain barrier [141]. This subacute phase offers a window for anti-inflammatory interventions [142], which may reduce secondary damage and improve functional outcomes.
Persistent Mitochondrial Impairment (Hours to Days): During this time, mitochondria exhibit impaired oxidative phosphorylation, sustained ROS production, and disrupted dynamics—including defective mitophagy and reduced biogenesis [143]. This prolongation of mitochondrial dysfunction supports the rationale for extended treatment with agents that enhance mitochondrial resilience and function beyond the immediate post-injury window.
Chronic Phase (Days to Weeks): Long-term consequences of neurotrauma include neurodegeneration, gliosis, axonal disconnection, and synaptic remodeling. Although this phase is less amenable to acute neuroprotective strategies, it represents a potential window for interventions promoting neuroplasticity, rehabilitation, and regenerative therapies aimed at functional recovery [144].
In summary, effective neurotrauma treatment hinges on recognizing and targeting the temporal dynamics of secondary injury. From rapid excitotoxicity and mitochondrial damage to inflammation and chronic degeneration, each phase offers a distinct therapeutic window. Tailoring interventions to these windows is crucial to maximizing neuroprotection and improving clinical translation.
We embedded relevant discussions throughout the manuscript, focusing on glutamate’s physiological role and its pathological contribution to excitotoxicity after neurotrauma, as well as therapeutic strategies.
Glutamate, the brain’s primary excitatory neurotransmitter, is essential for synaptic plasticity, learning, and memory. However, following TBI or SCI, excessive glutamate release and impaired astrocytic reuptake lead to toxic extracellular accumulation [74, 145]. This overactivates ionotropic (e.g., NMDA, AMPA, kainate) and metabotropic glutamate receptors, triggering calcium (Ca2+) and sodium (Na⁺) influx, oxidative stress, and mitochondrial dysfunction, ultimately leading to necrosis and apoptosis [11, 146]. Concurrently, activated microglia and astrocytes release inflammatory mediators, worsening neuronal damage [147].
While preclinical studies show that modulating glutamatergic signaling reduces neurotoxicity, clinical translation has proven difficult. NMDA receptor antagonists such as MK-801, memantine, and amantadine reduce excitotoxic damage in animal models [148, 149]. Amantadine demonstrated functional benefits in a TBI trial [86], yet broad NMDA inhibition often impairs normal brain function. More selective GluN2B-subunit antagonists (e.g., ifenprodil, traxoprodil) show better safety and efficacy in reducing edema and blood–brain barrier disruption in animal studies [150, 151, 152].
Targeting metabotropic glutamate receptors (e.g., mGluR5) has also shown promise; delayed inhibition post-TBI reduces neuronal loss and improves outcomes in rodents [153]. Additionally, drugs inhibiting glutamate release or ionic influx (e.g., lamotrigine, phenytoin, riluzole) offer neuroprotection by stabilizing neuronal membranes [154, 155]. Riluzole, in particular, has shown efficacy in SCI models and is under investigation for TBI [156].
Mitochondrial-targeted therapies, such as mPTP inhibitors (e.g., cyclosporin A) and antioxidants (e.g., edaravone, alpha-lipoic acid, CoQ10, melatonin), reduce ROS and preserve cellular energy production [157, 158, 159, 160]. Anti-inflammatory agents like minocycline and progesterone have demonstrated reduced microglial activation and cytokine release post-injury [161, 162, 163]. However, a Phase III trial of progesterone in TBI was halted for futility [164]. Other experimental therapies include erythropoietin, zinc, and endocannabinoid modulators, though results remain inconsistent [165].
Clinical translation faces several obstacles. Timing is critical—NMDA receptors are hyperactive shortly after injury but become desensitized within hours [166], necessitating early drug administration. In contrast, partial agonists like d-cycloserine show benefits when administered later [167], indicating that therapeutic windows vary with drug mechanism.
Biological factors such as sex and age also modulate excitotoxic vulnerability. Estrogen and progesterone confer neuroprotection, with women showing less oxidative stress and excitotoxic damage than men [168]. Aging increases susceptibility to glutamate-induced injury, as older brains exhibit higher oxidative stress and impaired resilience [169].
A persistent gap exists between animal models and human patients. Most preclinical studies use young, healthy rodents, which fail to represent the heterogeneity of human TBI/SCI, including age, sex, comorbidities, and complex injury profiles [170]. Additionally, the lack of real-time biomarkers for glutamate levels or receptor activity hinders precision treatment. CSF and serum markers such as glutamate or neurofilament are used in research but are not standard in clinical care [171]. Poor trial design—small sample sizes, heterogeneous populations, and lack of stratification—further impedes progress. For example, a large progesterone trial enrolled a wide glasgow coma scale (GCS) range (4–12), potentially diluting therapeutic effects [164].
To address these limitations, several strategies are emerging:
• Biomarker development (e.g., proton magnetic resonance spectroscopy, glial
fibrillary acidic protein (GFAP), neurofilament) for patient stratification and
therapeutic monitoring [172], • Combination therapies to target multiple injury mechanisms synergistically, • Precision medicine approaches tailored by sex, age, and genomic markers, • Improved preclinical models including aged, female, and comorbid animals, • Global health focuses on affordable interventions for low- and middle-income
countries (LMICs), such as magnesium sulfate, hypothermia, and early
rehabilitation [8], • Rigorous clinical trial designs with adaptive protocols and stratified
enrollment.
TBI affects approximately 69 million people annually worldwide, and SCI incidence continues to rise, particularly in LMICs [9, 173]. Addressing glutamate-driven secondary injury is a critical goal, but must be integrated with public health strategies such as prevention, early trauma care, and accessible rehabilitation. A multidisciplinary and globally-informed approach could significantly improve neurotrauma outcomes.
Glutamate excitotoxicity is a well-established yet persistently challenging mechanism underlying neuronal damage in neurotrauma, particularly in TBI and SCI. This study highlights the acute glutamate surge and NMDA receptor overactivation that lead to calcium-mediated cascades involving oxidative stress, mitochondrial dysfunction, and neuroinflammation—core drivers of secondary injury. Although preclinical models have identified multiple therapeutic targets within this cascade, clinical translation has largely faltered due to timing issues, limited efficacy, and adverse side effects. These findings emphasize a crucial gap between bench and bedside, revealing that single-pathway interventions may be insufficient in addressing the multifactorial nature of neurotrauma. Importantly, the research emphasizes the need for next-generation therapeutics with multi-modal actions—particularly those that concurrently modulate excitotoxicity and inflammation while maintaining a favorable safety profile. Future directions should prioritize combination therapies, better therapeutic windows, consideration of age and gender, and precision delivery methods. Clinically meaningful advances will require rethinking trial designs to reflect the complexity of neurotrauma pathology and those factors stated earlier. As the field progresses, continued inquiry is vital—not only to refine therapeutic strategies but also to deepen our understanding of glutamate’s role in CNS injury. A coordinated translational effort can ultimately lead to interventions that improve long-term neurological outcomes for patients with TBI and SCI.
JH and PB conceptualized and designed the study. JH, KK, and PB contributed to drafting and reviewing the manuscript. PB participated in editorial revisions, and KK prepared the figures. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Graphical abstract created in BioRender. Bose, P. (2025) https://BioRender.com/b0d5h8p.
Supported by Research Award # SC210266 and SC220248 from the United States Department of Defense (DoD), Merit Review Award # B3986-R/1 I01 RX003986-01A1, and SPiRE Award # B4097-P/I21 RX004097 from the United States Department of Veterans Affairs Research and Development Service (R&D).
The authors declare no conflict of interest.
During the preparation of this work, the authors used Grammarly and ChatGPT in order to check spelling and grammar.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.