


 , Asuka Kawachi 1, Akihide Yoshimi 1,*
, Asuka Kawachi 1, Akihide Yoshimi 1,*
1 Division of Cancer RNA Research, National Cancer Center Research Institute, 104-0045 Tokyo, Japan
Abstract
Clonal hematopoiesis (CH) is characterized by the expansion of hematopoietic stem and progenitor cells harboring somatic mutations, which confers an increased risk of hematologic malignancies and cardiovascular disease. Among CH-associated mutations, mutations affecting splicing factors (SFs), including splicing factor 3b subunit 1 (SF3B1), serine/arginine-rich splicing factor 2 (SRSF2), U2 small nuclear RNA auxiliary factor 1 (U2AF1), and zinc finger CCCH-type, RNA binding motif and serine/arginine rich 2 (ZRSR2), play a unique role in promoting clonal expansion and leukemogenesis. In this review, we summarize recent findings on the role of SF mutations in CH progression, their interplay with other mutations (e.g., DNA methyltransferase 3 alpha (DNMT3A), ten-eleven translocation methylcytosine dioxygenase 2 (TET2) and isocitrate dehydrogenase 2 (IDH2)), and their impact on hematopoietic homeostasis. Epidemiological studies have demonstrated that SF-mutant CH exhibits an accelerated clonal expansion compared to other CH clones. Furthermore, murine models suggest that SF mutations alone do not inherently confer a growth advantage for clonal expansion but rather enhance disease phenotypes when co-existing with epigenetic mutations, such as IDH2 and TET2. These findings suggest that SF mutations contribute to CH expansion and malignant transformation through a synergistic interplay with other mutations and external factors such as inflammation. Given the clinical significance of SF mutations, ongoing research is focused on developing targeted therapies that modulate aberrant RNA splicing and prevent CH-driven leukemogenesis. Understanding the mechanisms underlying mutant spliceosome-mediated CH expansion may provide novel insights into early detection, risk stratification, and therapeutic interventions in hematologic malignancies.
Keywords
- clonal hematopoiesis
- splicing factor
- mutation
- leukemia
CH is defined by the detection of somatic mutations in hematopoietic stem and progenitor cells (HSPCs), which have the capacity to expand over time under selective clonal pressures. A landmark study in 2014, which analyzed whole-exome sequencing data from 17,182 individuals, confirmed the high prevalence of CH in individuals over 40 years old [1]. Specifically, CH was detected in 9.5% of individuals aged 70–79, 11.7% in those aged 80–89, and 18.4% in individuals over 90 years old. Furthermore, CH was associated with an increased risk of hematologic malignancies, with a hazard ratio (HR) of approximately 10, indicating a substantially elevated risk of developing conditions such as acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS). Interestingly, mutations associated with CH predominantly occur in epigenetic regulatory genes such as DNA methyltransferase 3 alpha (DNMT3A), ten-eleven translocation methylcytosine dioxygenase 2 (TET2) and additional sex combs-like 1 (ASXL1), which are also frequently observed in myeloid neoplasms, suggesting a potentially pre-malignant state. In addition, mutations in splicing factor (SF) genes, including SF3B1, SRSF2, U2AF1, and ZRSR2, are detected at moderate frequencies [2]. The detection of a clonal cell population depends on both sequencing depth as well as the coverage and design of the sequencing platform.
Mutations in SF3B1, SRSF2, and U2AF1 typically occur at characteristic hotspot residues and are considered change-of-function mutations that confer abnormal or novel splicing activity. In contrast, mutations in ZRSR2 lack recurrent hotspots and mostly result in loss of function. This observation is consistent with the fact that ZRSR2 is located on the X chromosome, and MDS or chronic myelomonocytic leukemia cases harboring ZRSR2 mutations are more frequently observed in males [3].
Based on both clinical and molecular features, CH can be categorized into four major subtypes: aging-related clonal hematopoiesis (ARCH), clonal hematopoiesis of indeterminate potential (CHIP), idiopathic cytopenia of undetermined significance (ICUS), and clonal cytopenia of undetermined significance (CCUS) (Table 1 (Ref. [1, 4, 5])). Each entity exhibits distinct clinical and prognostic implications.
| Clonality (VAF %) | Dysplasia | Cytopenias | BM blast % | Progression to MN | Median Age (Range) | Ref | |
| ARCH | generally |
– | – | Not well established | |||
| CHIP | – | 0.5% per year | 57 (44–73) | [1] | |||
| ICUS | + | 5 years C.I. | 53 (18–88) | [4] | |||
| 9.0% | |||||||
| CCUS | + | 2 years C.I. | 70 (19–94) | [5] | |||
| 12.6% | |||||||
| 5 years C.I. | 68 (18–86) | [4] | |||||
| 82.0% |
VAF, variant allele frequency; ARCH, aging-related clonal hematopoiesis; CHIP, clonal hematopoiesis of indeterminate potential; ICUS, idiopathic cytopenia of undetermined significance; CCUS, clonal cytopenia of undetermined significance.
ARCH refers to the age-related acquisition of somatic mutations in hematopoietic cells that results in clonal expansion without evident clinical consequences. Unlike CHIP, ARCH-associated mutations typically occur at lower variant allele frequencies (VAF) and do not necessarily confer a significantly increased risk of hematologic malignancies. ARCH is considered a natural byproduct of aging and genomic instability.
CHIP is defined by the presence of somatic mutations in genes commonly
associated with hematologic malignancies in individuals without cytopenias or
dysplastic hematopoiesis. The VAF is typically
ICUS is a condition characterized by persistent, unexplained cytopenias that do not meet the diagnostic criteria for MDS or other hematologic malignancies. Unlike CCUS, ICUS is not necessarily associated with detectable clonal mutations, and its etiology remains uncertain. Some cases of ICUS may evolve into clonal disorders over time.
CCUS is defined as the coexistence of cytopenia and clonal hematopoiesis without fulfilling the criteria for MDS or other hematologic neoplasms. It is distinguished from CHIP by the presence of cytopenias and from ICUS by the presence of identifiable somatic mutations. Individuals with CCUS exhibit a significantly higher risk of progression to MDS or AML compared to CHIP.
These classifications help delineate the varying degrees of risk associated with CH and provide a framework for monitoring individuals with clonal hematopoiesis. Further research is needed to refine the prognostic implications and determine optimal management strategies for each subtype.
RNA splicing is a crucial process in eukaryotic gene expression, whereby introns are removed from the primary transcript, and the remaining exons are precisely ligated to generate a mature mRNA. SFs play essential roles in facilitating and regulating this process, ensuring the proper removal of introns and exon-exon junction formation. Mutations in genes encoding splicing-related factors can disrupt splicing fidelity, leading to aberrant splicing events such as exon skipping, intron retention, and the utilization of cryptic splice sites. These dysregulated splicing events result in widespread alterations in transcript isoforms, significantly impacting protein expression and function. Consequently, they contribute to tumorigenesis and chronic inflammatory conditions.
Among the various reported mutations in SF genes, the majority are concentrated in four key genes: SF3B1, SRSF2, U2AF1, and ZRSR2. Notably, SF3B1 mutations are detected in 60–70% of MDS with ring sideroblasts, making it one of the most frequently mutated genes in this subtype [6]. When considering all subtypes of MDS, SF mutations are identified in approximately 50% of cases. Additionally, SRSF2 mutations are found in 40–50% of chronic myelomonocytic leukemia (CMML) cases, further underscoring the strong association between SF mutations and hematologic malignancies [7].
Although specific patterns of SF mutations are linked to distinct disease subtypes, these mutations are widely observed across various hematologic neoplasms. In this review, we provide an overview of the pathophysiological mechanisms by which SF mutations contribute to the development of CH and their clinical significance.
The development of CH is driven by specific driver gene mutations that play a crucial role in facilitating clonal expansion. Among these, DNMT3A, TET2, and ASXL1 (DTA) are the most frequently mutated genes in CH. These mutations accumulate in an age-dependent manner and promote clonal expansion once a critical threshold frequency is reached. In addition to DTA mutations, spliceosome mutations, including SRSF2, SF3B1, U2AF1, and ZRSR2, exert distinct effects on CH progression. Notably, hotspot mutations such as SRSF2P95H, SF3B1K700E, and U2AF1S34F induce specific mRNA splicing abnormalities, disrupting hematopoietic homeostasis. These alterations confer a selective advantage to CH clones and are implicated in an elevated risk of progression to myeloid malignancies. A recent study has further demonstrated that in CHIP, SF gene mutations (e.g., SF3B1, SRSF2, ZRSR2) represent the second most frequently observed class of mutations, following DTA [8].
In some cases of CHIP, decelerated growth of certain clones has been observed, suggesting that CH clones with lower fitness may undergo natural extinction with aging [9, 10]. A 21-year longitudinal study conducted on a general population cohort from the Atherosclerosis Risk in Communities study, with a median enrollment age of 55 years, demonstrated that SF mutations (SF3B1, SRSF2, U2AF1, ZRSR2) and TET2 mutations exhibited significantly faster clonal expansion compared to other CHIP-associated mutations [10]. Clones harboring DNMT3A or TP53 mutations exhibit an annual growth rate of approximately 5%, whereas clones carrying SF3B1, U2AF1, and non-P95H SRSF2 mutations show a more rapid expansion rate of 10–20%. Remarkably, clones carrying the SRSF2 P95H mutation demonstrated an exceptionally high growth rate of over 50% [9]. Furthermore, CH clones with a high growth rate are associated with an increased risk of progression to AML. Clones harboring SRSF2 or U2AF1 mutations are strongly linked to a higher AML risk, highlighting the clinical significance of these mutations in leukemogenesis.
A real-world data study involving 357 patients with CCUS demonstrated that SRSF2 mutations (n = 61, 10.3%) were the third most frequently observed, following TET2 and DNMT3A mutations. Additionally, U2AF1, SF3B1, and ZRSR2 mutations were identified with lower prevalence, following ASXL1 mutations [5].
A previous study has reported that when CH screening is performed on individuals
with unexplained cytopenia, CH-related mutations are detected in approximately
45–60% of patients, a condition referred to as CCUS. Furthermore, it has been
demonstrated that the presence of SF mutations is associated with a significantly
increased risk of developing myeloid neoplasms, with a positive predictive value
of 0.8–1.0 [4]. Additionally, a study reported that older adults (
Emerging evidence suggests that inflammatory and immune-mediated stress exerts
selective pressure on hematopoietic clones, particularly those bearing mutations
that confer resistance to cytokine-driven apoptosis or differentiation. Clones
with loss-of-function mutations in Tet2 have been shown to expand under
chronic exposure to pro-inflammatory signals such as TNF-
Despite the frequent detection of SF mutations such as SRSF2, SF3B1, and U2AF1 in clonal hematopoiesis and secondary AML, their interaction with inflammatory signaling remains poorly characterized. However, the precise mechanisms by which individual cytokines influence the behavior of SF-mutant clones have yet to be elucidated. It is conceivable that, as with DTA, SF-mutant clones may exploit chronic inflammation to persist and expand. Further experimental studies are needed to delineate the causal relationship between cytokine-mediated stress and SF-mutant clonal fitness, which may offer new insights into the pathogenesis of clonal hematopoiesis.
Not all individuals with CH develop myeloid neoplasms, and the precise incidence remains uncertain due to the lack of rigorous prospective data. However, for example, the risk of CHIP progressing to AML is estimated to be 0.5–1.0% per year [17]. Considering that the prevalence of CHIP in 70-year-old individuals is over 100 times higher than that of MDS or AML, it is believed that the majority of individuals with CHIP do not develop hematologic malignancies [18]. In contrast, CCUS is known to have a significantly higher progression rate to myeloid neoplasms. The 5-year and 10-year cumulative progression probabilities were 82% and 95% for CCUS, compared to 9% and 9% for ICUS, respectively, demonstrating a much higher risk in CCUS. A real-world study involving 357 CCUS patients showed that SF mutations significantly increased the risk of progression from CCUS to myeloid neoplasms. Specifically, the leukemia-free survival (LFS) was significantly shortened in patients with SRSF2 (HR, 3.81; 95% CI, 2.13–6.83; p = 0.001) and ZRSR2 mutations (HR, 3.19; 95% CI, 1.43–7.12; p = 0.002) [5]. Additionally, a single-center study of 24 CH patients with U2AF1 mutations reported that 23 patients (96%) met the criteria for U2AF1-mutant CCUS, and 6 patients (25%) progressed to MDS or AML within the median time to transformation of 17.5 months, although it remains unclear how many of these progression cases met the formal criteria for CCUS [19].
The risk of developing myeloid neoplasms is associated with the type of somatic
mutations present in CH. A study analyzing pre-diagnostic samples from 95
individuals who subsequently developed AML (pre-AML group) with an average
follow-up of 6.3 years, found that driver mutation-positive ARCH was
significantly more prevalent in the pre-AML group compared to controls (73.4%
vs. 36.7%, p
Recent efforts to model CH in animal models have aimed to explore the relationship between CH and genetic mutations. Mutations associated with CH initially arise in a small subset of HSPCs and gradually expand over time [21]. This implies that CH clones must possess a proliferative advantage over normal hematopoietic clones in vivo. To model this process, competitive bone marrow transplantation has been employed.
For example, when Tet2-deficient bone marrow cells (10%) were
transplanted along with 90% wild-type bone marrow cells, the
Tet2-deficient cells gradually expanded in the bone marrow, spleen, and
blood, demonstrating a mild myeloid bias with preferential expansion in
Ly6Chigh monocyte subsets [22]. Similarly, Dnmt3a-deficient
hematopoietic stem cells exhibited clonal expansion when transplanted
competitively, particularly under conditions of chronic mycobacterial infection
or IFN-
Mouse models have also been developed to investigate the impact of SF mutations. Mupo et al. [23] introduced the Sf3b1K700E mutation into mouse HSCs and performed competitive transplantation with wild-type cells. While Sf3b1K700E mutant cells initially showed good engraftment at one-month post-transplantation, by four months, leukocyte production from Sf3b1K700E/+ cells was significantly reduced compared to wild-type cells, suggesting that the Sf3b1K700E mutation negatively affects the long-term self-renewal capacity of HSCs. This phenomenon was observed in both young and aged recipient mice [23]. Similarly, HSCs derived from Srsf2P95H mutant mice exhibited impaired long-term reconstitution capacity compared to controls [24, 25]. These findings indicate that, to date, no animal model has successfully demonstrated a proliferative advantage of CH clones driven by SF mutations alone.
However, animal models have provided evidence that co-mutations with epigenetic regulators may influence CH phenotypic acquisition. Yoshimi et al. [26] generated mouse models that express both Srsf2P95H and Idh2R140Q mutations specifically within hematopoietic lineages. Competitive transplantation of mutant bone marrow (1:1 ratio) with wild-type bone marrow into lethally irradiated mice revealed that co-mutant cells exhibited a stronger proliferative advantage compared to IDH-mutant-only cells. Notably, these co-mutations led to a MDS-like phenotype with proliferative features and significantly reduced survival [26]. Similarly, in Srsf2P95H/+ Tet2-/- mice, no proliferative advantage over wild-type HSCs was observed in competitive transplantation assays. However, these mice exhibited a CMML-like phenotype, underscoring the impact of co-mutations in driving disease progression [27].
SF3B1 mutations contribute to aberrant splicing through a distinct mechanism. These mutations are enriched in the HEAT (Huntingtin, Elongation factor 3, protein phosphatase 2A, TOR1) repeat domain, which is not directly involved in RNA binding but is rather implicated in protein-protein interactions within the U2 snRNP complex [3, 28, 29]. In canonical splicing, SF1 binds to the branchpoint (BP) sequence with the assistance of U2AF, facilitating accurate BP recognition (Fig. 1A). Subsequently, the U2 snRNP is recruited, and SF3B1, via its HEAT repeat domain, recruits SUGP1 [30]. SUGP1 directly interacts with both SF1 and U2AF2, helping to position U2 snRNP correctly near the canonical BP and 3′ splice site (3′ss). The G-patch domain of SUGP1 interacts with and activates a DEAH-box RNA helicase, which in turn displaces SF1 via the action of p14, enabling base pairing between U2 snRNA and the canonical BP sequence. However, when SF3B1 carries cancer-associated mutations in its HEAT domain, the recruitment of SUGP1 is impaired, and consequently, its ability to recruit and activate the RNA helicase is reduced [31]. In this context, other G-patch domain-containing proteins, such as GPATCH8, can outcompete SUGP1 for helicase binding [32]. As a result, the helicase activation is compromised, SF1 remains bound, and access to the canonical BP is blocked. This failure forces U2 snRNP to utilize upstream BPs and cryptic 3′ss, a splicing phenotype characteristic of tumors with mutant SF3B1 [30, 31].
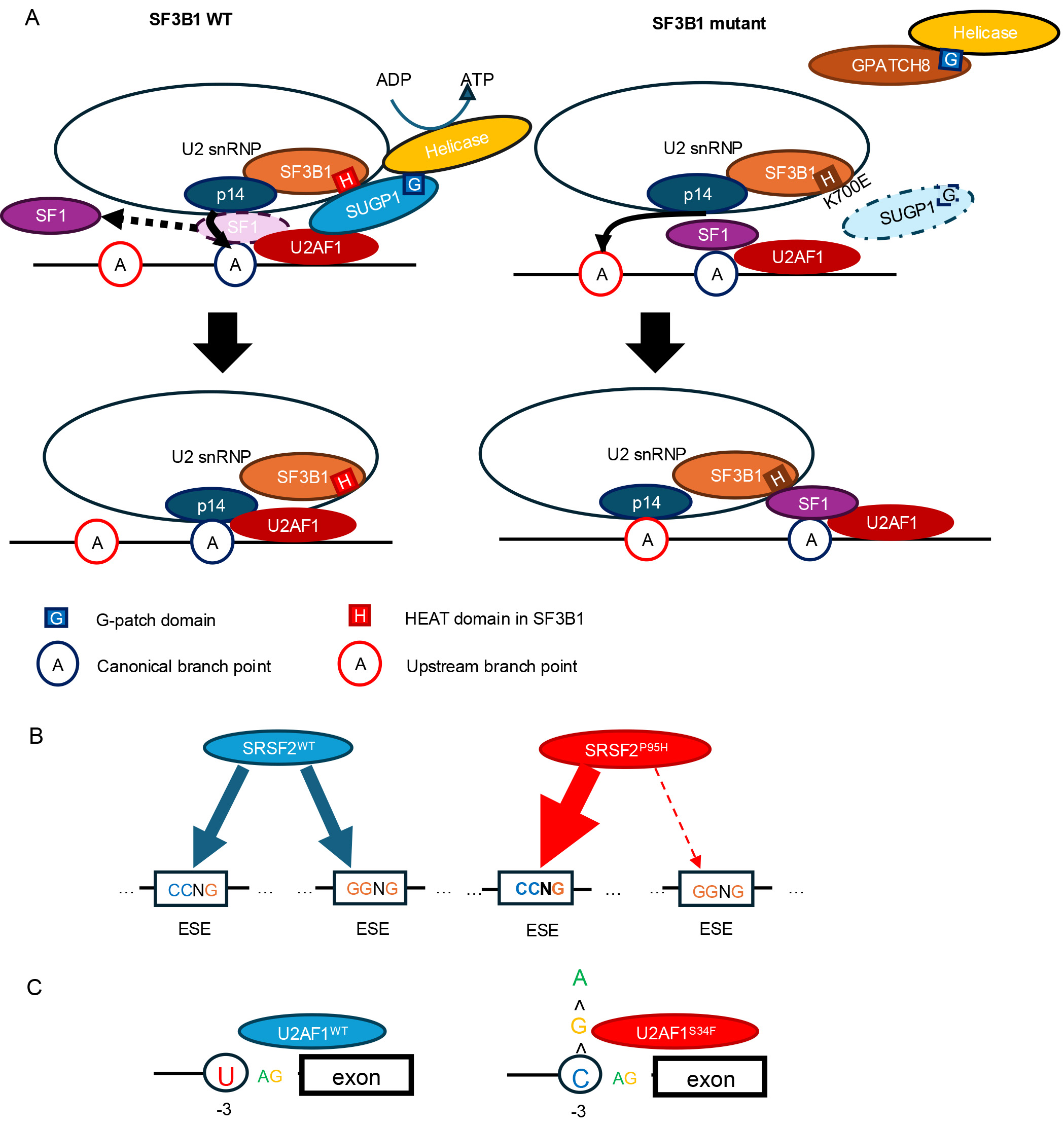 Fig. 1.
Fig. 1.
Mechanisms by which major splicing factor mutations alter pre-mRNA splicing. (A) SF3B1K700E impairs recruitment of SUGP1, preventing displacement of SF1 and allowing p14 to recognize an upstream branch point. (B) SRSF2P95H skews the equal recognition of CCNG and GGNG motifs, shifting the preference toward CCNG sequences (ESE, exonic splicing enhancer). (C) U2AF1WT preferentially recognizes the sequences containing U at the –3 position relative to the 3′ splice site, whereas U2AF1S34F shifts this preference toward sequences containing C at the same position. SF, splicing factor.
SF3B1 mutations have been reported to contribute to disease phenotypes
and tumorigenesis through multiple cellular pathways (Fig. 2A). Specifically,
SF3B1 mutations have been shown to induce mis-splicing of
MAP3K7 and PPP2R5A genes [33, 34]. In both cases,
SF3B1 mutations promote the usage of cryptic 3′ splice sites,
leading to nonsense-mediated decay (NMD). Reduced expression of MAP3K7 results in
the activation of the NF-
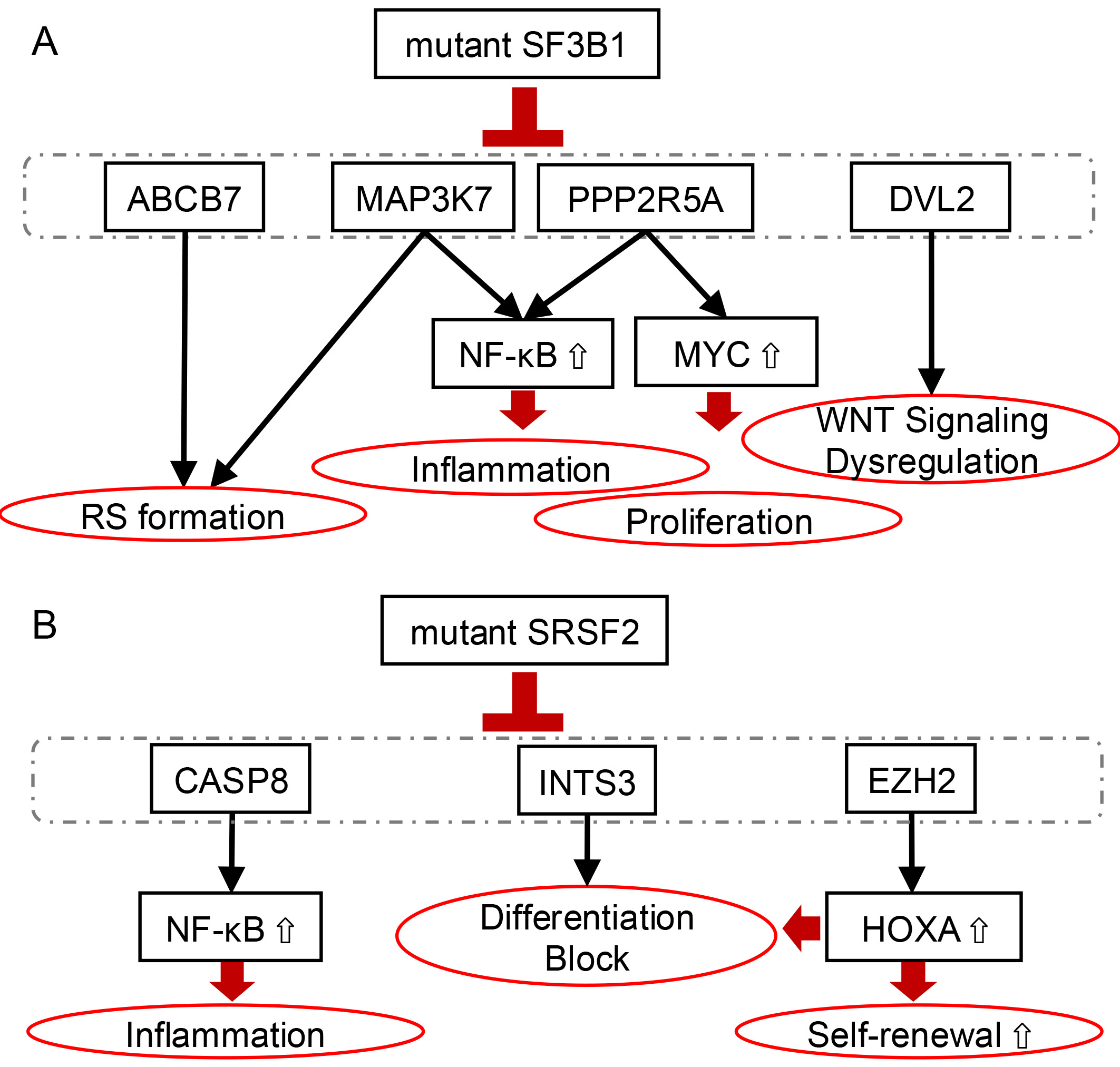 Fig. 2.
Fig. 2.
Oncogenic pathways induced by mutant SF3B1 and SRSF2 in clonal
hematopoiesis and myeloid malignancies. (A) Mutant SF3B1 induces aberrant
splicing of critical regulatory genes, including ABCB7,
MAP3K7, pPP2R5A, DVL2, and RNH1. These
alterations lead to enhanced proliferation and inflammation, dysregulation of WNT
signaling, and ring sideroblast (RS) formation. (B) Mutant SRSF2 causes
mis-splicing and downregulation of CASP8 and EZH2, resulting in enhanced
NF-
SRSF2 regulates splicing by binding to exon splicing enhancer (ESE) sequences within pre-mRNA and physically interacting with U1 and U2 snRNPs [36, 37, 38]. The hotspot mutation in SRSF2 occurs at proline 95. Wild-type SRSF2 binds C-rich and G-rich RNA motifs (such as GGNG and CCNG) with similar affinity. In contrast, mutant SRSF2 preferentially recognizes C-rich sequences, thereby promoting the inclusion of exons harboring C-rich ESE and altering splicing patterns (Fig. 1B) [24, 39]. Because the SRSF2P95H mutation occurs within a GC-rich sequence context, it is prone to underdetection due to reduced sequencing coverage. Yoshimi et al. [26] reanalyzed the entire RNA-seq dataset from the TCGA AML cohort (n = 179), which had initially identified only one SRSF2 mutation-positive case, and were able to identify 19 cases (11%) with SRSF2 mutations by detecting a characteristic mis-splicing pattern involving 70 genes associated with SRSF2 mutations. In particular, the abnormal inclusion of a “poison exon” containing a premature stop codon within the EZH2 transcript results in the mis-splicing of EZH2, leading to NMD and subsequent degradation of EZH2 transcripts, thereby reducing EZH2 protein expression (Fig. 2B). Similarly, aberrant splicing of INTS3 induced by mutant SRSF2 leads to intron retention and exon skipping, resulting in NMD-mediated degradation of INTS3 transcripts. This downregulation of INTS3 impairs the integrator complex and contributes to defective myeloid differentiation and enhanced leukemogenic potential.
Mutations in SRSF2 promote aberrant splicing of the CASP8, leading to
exclusion of a cassette exon (Fig. 2B) [25]. This results in the expression of a
C-terminally truncated Caspase-8 isoform that lacks the catalytic domain but
retains the N-terminal prodomain. This truncated form is incapable of inducing
apoptosis, but it hyperactivates NF-
SRSF2 mutations are also known to induce aberrant splicing of transcription factors such as BCOR, as well as RNA-binding proteins belonging to the hnRNP and SR protein families [24, 40]. Liang et al. [40] demonstrated that the SRSF2P95H induces exon skipping in HNRNPA2B1 transcripts, leading to impaired differentiation of human CD34⁺ HSPCs. Additionally, the SRSF2P95H mutation is known to stabilize PINK1 mRNA, leading to an increase in PINK1 protein expression [41]. As a result, cells harboring SRSF2 mutations exhibit enhanced dependence on mitophagy, conferring a survival advantage.
Despite their distinct splicing alterations, mutations in SF3B1 and
SRSF2 both converge on aberrant activation of the NF-
The U2AF1/U2AF2 heterodimer recognizes the AG dinucleotide at the 3′ss of precursor mRNA within introns. In this complex, U2AF2 binds to the polypyrimidine tract within the intron as well as to SF3B1, while U2AF1 directly interacts with the AG dinucleotide at the exon–intron junction. Mutations in U2AF1 occur within one of its two zinc finger domains, and RNA-seq analyses in U2AF1-mutant cells consistently show that these mutations alter splicing patterns depending on the nucleotide context flanking the 3′ss (Fig. 1C) [42]. Specifically, S34 mutations affect the –3 position upstream of the AG, such that exons with a U at the –3 position are more likely to be skipped, while those with a C at –3 are preferentially included. In contrast, Q157 mutations influence the +1 position, immediately downstream of the AG dinucleotide. Exons with a G at the +1 position tend to be included, whereas those with an A at that site are more frequently skipped.
Cells harboring the U2AF1 S34F mutation exhibit a significant increase in
reactive oxygen species prodution [42]. Both mitochondrial and peroxisomal
functions are impaired, leading to the accumulation of hydrogen peroxide.
Concurrently, the expression of key DNA damage response proteins, such as ATM,
ATR, and CHEK1/2, is downregulated, resulting in defective DNA repair processes.
This is evidenced by persistent phosphorylation of
CH is driven by somatic mutations in HSPCs, with an increased risk of
hematologic malignancies and cardiovascular disease. Among CH-related mutations,
SF mutations, particularly in SF3B1, SRSF2, U2AF1, and
ZRSR2, play a crucial role in clonal expansion and leukemogenesis. SF
mutations drive aberrant RNA splicing, altering gene expression and hematopoietic
homeostasis. A key challenge in understanding SF-mutant CH lies in the
discrepancy between human CH and murine models, as mouse models of
Sf3b1, Srsf2, and U2af1 mutations fail to demonstrate
a clear clonal advantage. This suggests that SF-mutant CH clones require
additional intrinsic or extrinsic factors to sustain expansion. Single-cell
co-mutational profiling of CH clones with SF mutations may provide deeper
insights into these dependencies. Furthermore, external stressors, such as
chronic infections and IFN-
TI wrote the manuscript. TI and AK contributed to the literature search. AY contributed to the design of the article structure, the exploration of innovative points, the revision and correction of drafts and funding. All authors contributed to editorial changes in the manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work. All authors read and approved the final manuscript.
Not applicable.
Not applicable.
This research was partially supported by grants awarded to AY from the Japan Agency for Medical Research and Development (AMED) (grant numbers JP24gm7010010h0001, 24ck0106906h0001, and 24ck0106946h0001) and the Takeda Science Foundation.
The authors declare no conflict of interest.
During the preparation of this work, the authors used ChatGpt in order to check spelling and grammar. After using this tool for the entire manuscript, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.