

 , Hadas Elalouf 2, Hanan Maoz 1
, Hadas Elalouf 2, Hanan Maoz 11 Health Management Program, Department of Management, Bar-Ilan University, 5290002 Ramat Gan, Israel
2 Information Science Department, Bar-Ilan University, 5290002 Ramat Gan, Israel
Abstract
This review provides a comprehensive analysis of recent advancements in elucidating the molecular mechanisms underlying human immunodeficiency virus (HIV)-1 entry, focusing on the intricate interplay between the viral envelope glycoproteins (Env) and host cell receptors. We detail how structural insights into glycoprotein (gp)120-Cluster of Differentiation 4 (CD4)/coreceptor interactions and gp41-mediated membrane fusion inform therapeutic interventions, including fusion inhibitors and broadly neutralizing antibodies (bnAbs). The HIV-1 Env trimer undergoes a series of highly coordinated conformational transitions from a metastable prefusion state to a stable postfusion structure. CD4 engagement induces allosteric remodeling of gp120, unveiling coreceptor (C-C chemokine receptor type 5 (CCR5)/C-X-C chemokine receptor type 4 (CXCR4)) binding sites and priming gp41 activation. Fusion peptide insertion, six-helix bundle formation, and membrane merger are critical targets for inhibitors like T20 (enfuvirtide). Comparative analyses with other viruses reveal conserved fusion mechanisms despite distinct activation triggers, offering broader insights for antiviral development. By integrating structural biology, virology, and translational research, this review highlights how the mechanistic dissection of viral entry informs the design of next-generation therapeutics. We highlight strategies to disrupt Env-receptor interactions, block fusion intermediates, and harness cross-viral principles to counteract drug resistance and refine vaccine approaches. These insights not only deepen our understanding of HIV-1 pathogenesis but also drive the innovation of novel antiviral strategies.
Keywords
- HIV-1 entry mechanisms
- protein-host receptor interactions
- membrane fusion dynamics
- viral envelope glycoproteins
Human immunodeficiency virus (HIV) primarily targets CD4+ T cells, a type of immune cell crucial for immune function. Untreated HIV infection progressively destroys these cells, leading to severe immune damage and eventually acquired immunodeficiency syndrome (AIDS) [1, 2]. AIDS drastically increases susceptibility to opportunistic infections and AIDS-defining cancers such as Kaposi’s sarcoma and non-Hodgkin lymphoma [3]. As of 2023, approximately 39.9 million people are affected by the global impact of HIV/AIDS, with 65% of the infections occurring in sub-Saharan Africa. Since its discovery, about 42.3 million deaths have been reported worldwide, particularly among women of reproductive age [4, 5, 6]. In 2023, approximately 1.3 million new HIV infections and 630,000 HIV-related deaths were confirmed [7]. In 2021, Israel reported 367 new HIV cases, with an incidence rate of 3.9 per 100,000, slightly up from 362 cases in 2020. By the end of 2021, 8386 individuals were living with HIV/AIDS, while cumulative deaths reached 1746 since the epidemic’s onset [8].
HIV primarily enters cells through interactions with specific receptors on the CD4 T cells. This process involves several steps, including binding to the CD4 receptor and a conformational change that enables the virus to fuse with the cell membrane and enter the cell [9, 10, 11]. HIV entry into host cells relies on the envelope glycoproteins (gp)120 and gp41. gp120 binds to the CD4 receptor on T cells, undergoes conformational changes, and then engages C-C chemokine receptor type 5 (CCR5) or C-X-C chemokine receptor type 4 (CXCR4) coreceptors, initiating the entry process. CCR5 is used by HIV strains that infect T cells and macrophages (R5 viruses), while CXCR4 is associated with later-stage infections (X4 viruses) [12, 13, 14]. The coreceptor binding induces structural rearrangements in gp41, facilitating viral envelope fusion with the host membrane and enabling viral RNA entry. The high variability and glycosylation of gp120 help HIV evade immune detection, enhancing infection efficiency [10, 15].
In the trimeric HIV envelope glycoprotein (Env) complex, a single CD4 molecule
interacts with a quaternary surface formed by two CD4-binding sites, one from the
outer domain of one gp120 protomer and another from the inner domain of an
adjacent gp120 protomer. This interaction stabilizes CD4-Env envelope
glycoproteins binding and is crucial for viral entry [16]. CD4 binding triggers
significant conformational changes in gp120, displacing the V1V2 loops by about
40 Å and exposing the coreceptor binding site [17]. The gp120 structure is
layered, with the gp41-interactive region maintaining the unliganded trimer’s
integrity while enabling gp120 flexibility for further conformational changes
[18]. Structural studies [16, 17, 18, 19, 20] have provided key insights into the molecular
dynamics of HIV-1 Env interactions with host receptors during viral entry.
Single-molecule Fluorescence (or Förster) resonance energy transfer (FRET)
studies revealed that the Env trimer transitions between three conformational
states—closed, intermediate, and open—with CD4 binding stabilizing the open
form [19, 20]. Crystal structures of gp120 in unliganded and CD4-bound states
identified a layered architecture that allows for conformational flexibility,
supporting both viral entry and immune evasion [18]. Cryoelectron tomograms
further characterized the mobility of the viral spike, suggesting that gp120’s
layered structure acts as a dynamic spacer facilitating interactions between the
outer domain and gp41 [18]. Additionally, chemical probe studies highlighted the
In a previous study, we conducted an in-depth analysis of the structural characteristics of HIV entry glycoproteins, such as gp160, gp120, and gp41, highlighting their roles in mediating viral entry into host cells [10]. The current review builds upon these findings and examines the molecular mechanisms governing the interaction between HIV entry proteins and host cell receptors, exploring how these interactions drive membrane fusion and facilitate efficient viral entry, ultimately contributing to a more comprehensive understanding of the viral invasion process.
The HIV envelope glycoprotein complex consists of gp120 and gp41, derived from a
precursor protein that forms a trimeric structure essential for viral entry.
gp120, the receptor-binding subunit, binds to the primary receptor CD4 on host
cells, while gp41, the fusion subunit, mediates viral and host membrane fusion
post-CD4 binding. In addition to CD4, HIV utilizes chemokine coreceptors (CCR5 or
CXCR4), influencing viral tropism. Host integrin
The Env glycoprotein of HIV-1, known as gp160, plays a crucial role in facilitating viral entry into host cells by mediating the fusion of the viral envelope with the host cell membrane. This glycoprotein is initially synthesized in the rough endoplasmic reticulum (RER) as a precursor, gp160, which cleaves into two subunits: gp120 and gp41. These subunits assemble into a trimeric spike on the virus surface, which is crucial for infectivity and immune evasion [10, 26]. The synthesis of gp160 involves several complex processes, including glycosylation, disulfide bond formation, and quality-control mechanisms to ensure proper folding. N-linked oligosaccharide modifications are added to proteins during their trafficking through the Golgi, where further processing occurs, contributing to the structural complexity and heterogeneity of the glycoproteins. Once cleaved by furin-like proteases in the Golgi, the mature gp120 and gp41 subunits remain loosely associated, forming functional viral spikes [27, 28].
Notably, the low incorporation of these spikes (approximately 10 per virion) on the virus surface aids immune evasion and limits cytopathicity. HIV-1 utilizes specialized structures known as virological synapses for efficient cell-to-cell transmission, a process in which Env plays a crucial role. These synapses enable the virus to propagate between cells, especially in T cells and macrophages, and help it evade immune detection during transfer [29, 30, 31]. The HIV-1 Env endures a multifaceted trafficking pathway essential to the virus’s infectivity. Synthesized in the endoplasmic reticulum (ER) as a 160-kDa precursor, gp160 forms trimers before being transported to the Golgi apparatus for further processing and maturation. Here, it undergoes proteolytic processing, yielding gp120 and gp41, which form trimeric spikes incorporated into virus particles at the plasma membrane. Env undergoes endocytosis through clathrin adaptors and can either be degraded or recycled back to the plasma membrane [27, 30, 31].
Structurally, gp160 comprises various domains, with gp120 containing variable and constant regions, while gp41 features fusion peptides and transmembrane (TM) domains (TMDs). Critical post-translational modifications include glycosylation and disulfide bonding. During Env’s processing, gp120 undergoes folding through distinct stages before its signal peptide cleaves, stabilizing the protein’s conformation [10, 26]. Key structural components, including the TMD and membrane-proximal external region (MPER), play crucial roles in viral entry by undergoing dynamic conformational changes from a prefusion to a post-fusion state. Cryo-electron microscopy (Cryo-EM) and nuclear magnetic resonance (NMR) studies have revealed these structures at lower resolutions, highlighting their importance in vaccine development and therapeutic targeting. Markedly, Env’s cytoplasmic tail (CT) modifications affect the antigenicity and fusogenic activity, with the CT interacting with Gag proteins during viral assembly [29, 32].
HIV-1 gp120 is integral to the HIV entry mechanism, functioning in tandem with the gp41 protein. It binds to the host CD4 receptor and coreceptors CCR5 or CXCR4, triggering viral fusion with the host cell. Beyond the entry, soluble gp120 induces apoptosis in cells like neurons via disrupted calcium homeostasis and oxidative stress, which leads to neuronal death [18, 27, 33, 34]. Structurally, gp120 consists of an inner and outer domain and a bridging sheet, which houses five conserved regions (C1–C5) and five variable loops (V1–V5). The variable regions contribute to immune evasion by altering glycosylation patterns, which shield gp120 from neutralizing antibodies. High-resolution studies reveal gp120’s conformational changes during CD4 binding, which is critical for coreceptor interaction and subsequent viral fusion [10, 26]. Glycosylation and disulfide bridges are crucial for the stability and function gp120, with N-linked glycans playing a key role in immune evasion. Structural studies also highlight the conserved regions responsible for CD4 binding and the role of the V3 loop in coreceptor specificity [35, 36, 37, 38].
The extensive glycosylation of HIV-1 gp120 serves as both a dynamic immune evasion mechanism and a modulator of viral entry efficiency, with recent studies revealing subtype-specific glycosylation strategies that balance these functions. Approximately 50% of gp120’s molecular mass consists of N-linked glycans, forming a conformationally adaptable shield that masks conserved epitopes while enabling receptor binding plasticity [39, 40]. HIV-1 gp120 utilizes glycan microheterogeneity to create a steric hindrance around CD4-binding and co-receptor interaction sites, enabling breathing motions that transiently expose and conceal vulnerable epitopes and stabilizing alternative Env conformations through glycan-protein interactions. Recent analyses using electron-transfer/higher-energy collisional dissociation (EThcD)-stepped-collision energy HCD (sceHCD)-tandem mass spectrometry (MS/MS) identified 18 conserved N-glycosylation sites and 5 O-glycosylation sites across HIV-1 subtypes, with spatial arrangements facilitating coordinated glycan movement during viral entry. Notably, V1/V2 loop glycans (N156, N160) form mobile “glycan gates” that regulate access to the CD4-binding site, while high-mannose clusters at N332/N392 act as decoys for non-neutralizing antibodies and as anchors for broadly neutralizing antibodies (bnAbs), such as potently germline-targeting antibody 128 (PGT128) [41, 42].
Comparative studies [43, 44] of HIV-1 subtypes reveal evolutionary adaptations
in glycan placement, with circulating recombinant form (CRF)07_BC exhibiting
dense V4/V5 glycans (N406/N413) conferring enhanced resistance to potently
germline-targeting antibody (PG)9/PG16 bnAbs, clade C viruses displaying
truncated N301 glycans in the V3 loop to increase CCR5 binding affinity, and
clade B viruses incorporating sialylated complex glycans that reduce dendritic
cell capture. The CRF07_BC strain exhibits a particular dependence on C2-domain
glycans (N197/N289) for maintaining envelope integrity, with their removal
resulting in complete loss of infectivity, in contrast to clade B viruses, where
these sites are more dispensable. Critical glycosylation sites mediating immune
escape include N160 (V2), which shields
Metabolic labeling study indicate that oligomannose-rich glycoforms (e.g., the Joel R. Haynes-fast low (JR-FL) strain) enhance dendritic cell capture via mannose receptor binding, facilitating trans-infection while reducing antibody accessibility [45]. In contrast, sialylated variants exhibit reduced immunogenicity but maintain infectivity through improved electrostatic interactions with target cells [43]. The glycan shield further modulates viral entry by optimizing co-receptor binding, maintaining Env conformation for CCR5/CXCR4 engagement, restricting protease accessibility at cleavage sites, such as N88/N230, to limit furin-mediated processing, and regulating membrane fusion through V3 loop glycans (N301/N332) that control the timing of fusion peptide exposure. Notably, selective deglycosylation at N386 (C3) enhances 2G12 bNAb binding by 300%, underscoring the potential for strategic glycan removal to expose conserved epitopes for vaccine targeting [39].
Emerging vaccine strategies leverage glycosylation insights, including glycan-hole engineering via directed removal of non-essential glycans (e.g., N197/N289) to focus antibody responses, cell-line optimization using human embryonic kidney (HEK)293-derived gp120 vaccines to preserve natural glycan heterogeneity [40], and subtype-specific immunogen design tailored to predominant regional glycoforms (e.g., mannose-rich vaccines for clade C-endemic areas). These approaches address the evolutionary paradox in which HIV-1 maintains glycan plasticity for immune evasion while conserving essential glycosylation sites for viral fitness. This is exemplified by cross-clade bnAbs, such as potently germline-targeting antibody DM (PGDM)1400, which targets conserved glycopeptide epitopes, validating this structural vulnerability as a promising therapeutic target [41, 42].
A recent study on the HIV-1CH040 Env trimer demonstrated that partially open Env trimers facilitate efficient viral entry while conferring resistance to bnAbs targeting gp120’s V1/V2 loops and V3-glycan regions. This structural plasticity enables gp120 to adopt intermediate conformations, optimizing immune evasion and receptor engagement. Notably, CH040 Env resists V1/V2-directed bnAbs (e.g., PG9, PGT145) but remains susceptible to CD4-binding site (CD4bs) bnAbs such as VRC01, suggesting that gp120’s variable loops provide epitope shielding without rigidly stabilizing the trimer in a closed conformation [46].
The viral lipid envelope attaches to host cell membranes primarily through the
TM glycoprotein gp41. This 345-amino-acid subunit comprises three key regions: a
21-residue TMD, a 172-residue extracellular domain (ectodomain), and a
142-residue C-terminal segment. Within the ectodomain, crucial fusion elements
include a polyproline region, a hydrophobic fusion peptide at the N-terminus, two
heptad-repeat regions (HR1 and HR2), and a tryptophan-rich membrane-proximal
external region (MPER). These elements assemble into
The fusion peptide is initially hidden within the gp120-gp41 complex but becomes
exposed after gp120 binds to CD4 and coreceptors. This exposure triggers membrane
destabilization and the formation of fusion pores, allowing viral entry. The
six-helix bundle formed by HR1 and HR2 is critical for this fusion process. It
can be disrupted by peptides derived from HR2, such as enfuvirtide (T-20), used
in HIV-1 therapy despite its cost and low oral bioavailability. Neutralizing
antibodies like 2F5 and 4E10 target the MPER region [18, 49, 50]. Due to the high
conservation, the mutations in the TMD anchor region of gp41 can impact
Env-mediated fusion. Models of gp41 suggest it forms a single
The CT of gp41 contains lytic peptide regions (LLP1-3), which regulate fusion efficiency through interaction with host membranes and the viral core. SERINC (serine incorporator) proteins, particularly SERINC5, limit HIV-1 infectivity by modulating the viral Env and membrane fusion processes. The role of gp41’s CT in HIV-1 entry is complex and remains debated, with multiple models suggesting different membrane-spanning configurations. Mutations and palmitoylation in this region impact Env incorporation into virions, viral infectivity, and cell-surface expression [52, 53, 54].
The gp120 and gp41 interaction is central to the HIV entry, driven by noncovalent bonds between these two proteins within the trimeric Env spikes. When gp120 binds to the CD4 receptor on host cells, it undergoes a conformational change that disrupts its interaction with gp41, revealing coreceptor-binding sites. This course triggers structural rearrangements in gp41, transitioning from a prefusion to a post-fusion state, which is essential for forming the fusion pore, allowing viral entry [10]. Various studies have examined the domains involved in gp120-gp41 interactions, identifying the inner domain gp120 and the heptad repeat 1 region of gp41 as key interaction points. Amino acid substitutions, such as W596A and W610A, can disrupt this interaction, and certain broad neutralizing antibodies interfere with these dynamics. Nevertheless, the flexibility of Env trimers can impact the antibody-targeted epitopes exposure [18, 37, 52, 54].
The interaction between gp41 and the HIV-1 matrix (Gag) is also crucial, particularly during viral assembly and maturation. Gag facilitates Env packaging during budding and regulates the Env structure during entry [55]. A study has identified the importance of the gp41 C-terminal tail (CT) in binding to Gag, which prevents premature entry into immature particles. Additionally, mutations in the gp41CT have been associated with resistance to HIV protease inhibitors [56]. The HIV-1 Tat protein interacts with the envelope glycoprotein Env, particularly with gp120, inducing conformational shifts that enhance viral entry by facilitating binding to CD4 and coreceptors. A study on CH040 Env revealed that mutations in the gp41 HR1 and HR2 domains compensate for destabilizing alterations at the gp120-gp41 interface, thereby preserving viral fitness. Substitutions such as Q567R in HR1 and S614G in gp120’s V1V2 domain restore trimer integrity, highlighting convergent evolution in maintaining Env functionality. This underscores the cooperative role of gp120’s apex (V1V2) and gp41’s HR regions in stabilizing the trimer during conformational transitions [57]. Tat complexes with trimeric Env modulate viral entry via alternative pathways. Interactions between Tat and gp120 can alter viral tropism and help the virus evade neutralizing antibodies, adding complexity to the immune response against HIV [10, 50, 58].
CD4, a host cell surface glycoprotein receptor comprising four immunoglobulin-like domains (D1–D4), features an ectodomain that extends approximately 115 Å from the cell surface, with D1 situated at the farthest point from the membrane [59]. CD4 plays a crucial role in activating T cells and in the progression of HIV-1 infection. During T cell activation, it binds to major histocompatibility complex (MHC) class II molecules on antigen-presenting cells, facilitating the recruitment of p56lck and tyrosine kinase, which are key components in activating T helper cells and regulating the adaptive immune response. In HIV-1 infection, CD4 is the virus’s primary receptor, initiating viral attachment and forming the coreceptor binding site on gp120. This interaction aligns gp120 on the viral surface with the coreceptor, a seven-TM protein in the cell membrane, requiring conformational adjustments in CD4, gp120, or both [21].
CXCR4 and CCR5 were recognized in 1996 as coreceptors essential for HIV-1 entry. Both are chemokine receptors within the G-protein-coupled receptor (GPCR) family, characterized by 7-TM segments. Coreceptor selection plays a key role in determining viral tropism [60]. Typically, CCR5-tropic (R5) viruses drive initial transmission, whereas CXCR4-tropic (X4) viruses or dual-tropic (R5X4) variants often appear in later stages of infection [61, 62]. Each receptor has a seven-TMs helical structure, an N-terminal segment, three extracellular and three intracellular loops, and a C-terminal tail. Various studies have examined modified CXCR4 constructs with stabilizing alterations and T4-lysozyme fusions complexed with different ligands. Additionally, a CCR5 variant fused with rubredoxin has been investigated in complex with maraviroc, an HIV-inhibiting drug, as well as the chemokine CCR5-[5P7] C-C motif chemokine ligand 5 (CCL5) [21, 60, 63].
GPCR structures typically exhibit a topology characterized by a 7-TM helical
bundle [64]. According to the two-site model, the N-terminal domain of CXCR4 or
CCR5 forms chemokine recognition site 1 (CRS1), which interacts with the globular
core domain of chemokines, while the 7TM helices form chemokine recognition site
2 (CRS2), serving as the binding pocket for the N-terminal domain of chemokines
[65]. Although these structures provide valuable insights into the architecture
of CCR5 and CXCR4, as well as their interactions with various ligands, they do
not offer detailed molecular information on their function as HIV-1 coreceptors
[63, 66, 67]. Several other cell-surface receptors, such as dendritic
cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN), the
mannose-specific endocytosis receptor, heparan sulfate,
The interaction between HIV and host integrins, particularly
For successful viral entry, all key components involved in membrane fusion must engage in a precisely coordinated interaction. Recent progress in structural biology has shed light on the molecular intricacies of these interactions, providing a deeper understanding of the mechanisms driving the fusion process.
Evidence regarding the interaction of Env-CD4 has emerged from the structural
analysis of gp120 in complex with both a CD4-induced antibody, 17b, and 2D CD4.
The core of gp120 comprises inner and outer domains stabilized by a four-stranded
Recent study reported that CD4 activates the open conformation in the Env trimer of the stabilized soluble env trimer design (SOSIP) design [74]. The B41 SOSIP Env trimer structure—a group B variant—was resolved at 3.7 Å in complex with the CD4i 17b Fab and CD4, showing V1-V2 flip and dislocation. This structure reveals multiple conformational adjustments induced by CD4, including the exposure of the V3 loop, repositioning of the fusion peptide, formation of the bridging sheet, and structural rearrangements in gp41. The newly formed bridging sheet and exposed V3 loop are crucial in the coreceptor binding site [75]. Remarkably, the V1 and V2 stem regions must undergo a 180-degree rotation to mimic the conformation in the CD4-induced bridging sheet. A cryo-EM reconstruction at 5.2 Å resolution was conducted to evaluate the effect of CD4 alone on Env conformation [74]. At this resolution, the presence or absence of 17b Fab did not produce additional conformational changes, indicating that the 17b Fab does not independently influence the Env structure.
In the SOSIP trimer Env, only one CD4 molecule can attach to get stabilized by a disulfide link due to mutant 201C and 433C residues, preventing CD4-induced alterations and captured by PGT145 [76]. CD4 assumes a different orientation in this configuration than when three CD4 molecules are linked to the Env trimer [16]. Some studies suggest that an additional CD4-binding site (BS-2) may be formed on the inner domain of a neighboring gp120 protomer following the initial interaction between Env and CD4. The structure of the SOSIP trimer within this complex and its unliganded version shows minimal changes. Unique conformational states have been identified using the smFRET method for the asymmetrical trimer bound by one and three CD4 molecules, labeled as state 2 and state 3, respectively [77, 78]. Nonetheless, it is still uncertain whether the partially open conformation observed when the gp120-gp41 interface-specific antibody 8ANC195 binds to the CD4-bound SOSIP trimer truly represents an intermediate stage in the natural opening process of the native Env [79, 80].
The 3.7-Å cryo-EM structure of the HIV-1 Env trimer complexed with
CD4-mimetic compounds BNM-III-170/M48U1 and the CD4-induced antibody 17b [81]
provides critical insights into the structural choreography of membrane fusion
initiation, revealing three key transitional states in the CD4-bound open
conformation that coordinate asymmetric protomer activation and gp41
restructuring for viral entry. The CD4-mimetic-bound Env exhibits a 15°
rotational displacement of gp120 protomers from the closed state, generating a 40
Å-wide trimer cavity for coreceptor engagement, a 40 Å translocation of
the V1V2 loop from the trimer apex to lateral positions adjacent to CD4-binding
sites [79], and the assembly of a four-stranded bridging sheet
(
The binding of BNM-III-170/M48U1 to the gp120 Phe43 pocket induces an 8 Å
inward displacement of the V1V2 base (residues 131–137), stiffens the V3 loop
via hydrogen bonding at N301/N332, and transmits allosteric signals through
A stepwise fusion model integrating cryo-EM data suggests that CD4 engagement
induces the collapse of the Phe43 pocket and displacement of the V1V2 loop
[79, 81], followed by asymmetric protomer opening, sequential coreceptor binding,
compaction of the HR1 helix leading to partial exposure of the fusion peptide,
and insertion of a sulfotyrosine at CCR5, triggering complete gp41 restructuring
and fusion pore formation. These structural insights present new therapeutic
avenues, where CD4-mimetic drugs could trap Env in semi-open states that are
vulnerable to V2i antibodies, HR1-stabilizing peptides could block helical bundle
formation (IC50
Efforts to crystallize a complex between the membrane-embedded coreceptor and Env have proven challenging. However, cryo-EM has overcome this barrier [84]. Previous studies [75, 78] have suggested that the coreceptor footprint on gp120 would involve both the bridging sheet and the V3 loop . It has been suggested that gp120 interacts with the coreceptor through the N-terminus and extracellular loop 2 (ECL2) of CCR5, as well as the N-terminus, ECL2, and ECL3 of CXCR4 [85, 86]. Additionally, a pair of V3 loops inserted into the 7-TM helices of the coreceptor’s CRS2 is proposed to be in direct contact with the gp120 bridging sheet, specifically at the N-terminus of the coreceptor [75].
Free energy calculations and molecular dynamics simulations have
been employed to model the interactions between the V3 loop and CCR5/CXCR4
[67, 87, 88]. It has been observed that enhanced HIV-1 entry can only occur
through tyrosine sulfation of the N-terminus of CCR5 but not for CXCR4 [89].
While cell signaling is facilitated by the C-terminal tail of CCR5, which
contains palmitoylation motifs and numerous phosphorylation sites, its function
as an HIV-1 coreceptor does not appear to rely on the C-terminal tail [90].
Several chemokines, including C-X-C motif chemokine ligand 12 (CXCL12)/stromal
cell-derived factor 1 (SDF-1), macrophage inflammatory protein-1 alpha
(MIP-1
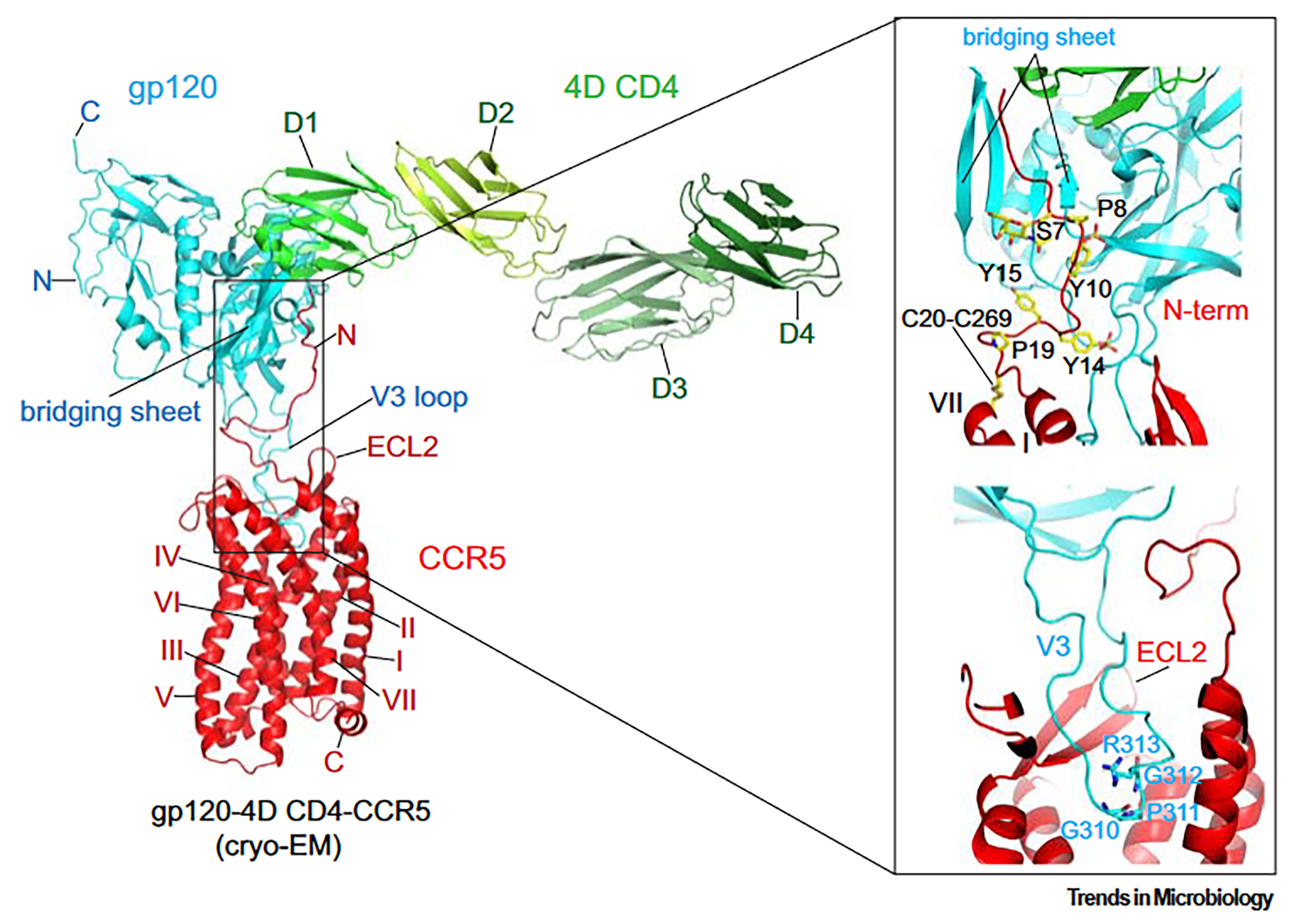
Cryo-EM has finally provided an intact and fully glycosylated structure of the gp120 complex with an unmodified human CCR5 and 4D CD4 [84]. Consistent with previous hypotheses [63], gp120 and CCR5 interact at two major interfaces (Fig. 1, Ref. [21]). Firstly, the V3 loop of gp120 inserts into CCR5’s CRS2 and directly contacts all 7-TM helices. At the tip of the V3 loop, the conserved 310GPGR(Q)313 motif penetrates approximately one-third of the lipid bilayer’s thickness into the CRS2 pocket (Fig. 1), with Pro311 being the most deeply embedded residue. Interestingly, a region near the V3 tip in the N-terminus of [5P7] CCL5 adopts a conformation similar to that of a corresponding segment in gp120. Proline residues in both structures—Pro311 in gp120 and Pro3 in [5P7] CCL5—extend to the bottom of the CRS2 pocket (Fig. 1).
 Fig. 1.
Fig. 1.
Interaction between envelope glycoprotein (Env) and CCR5. Left:
The overall structure of the CD4-gp120-CCR5 complex (PDB ID: 6MET) is depicted
as a ribbon diagram, highlighting the N- and C-termini, ECL2, and TM helices
I–VII. The V3 loop and the bridging sheet of gp120 are also labeled. Right:
Detailed views of the gp120-CCR5 interaction reveal the N-terminal region of CCR5
engaging with the four-stranded
The interaction between CCR5 and gp120 forms a semicircular grip, with the ECL2 of CCR5 encircling the V3 loop and making direct contact with residues from the crown and V3 stem. The N-terminus of CCR5 and the bridging sheet gp120 form the second key interaction interface between Env and its coreceptor. The N-terminal region of CCR5 assumes an extended structure, interacting with the surface of the bridging sheet through multiple abrupt turns (Fig. 1). Within CCR5, three tyrosine residues—Tyr15, Tyr14, and Tyr10—are sulfated, with Tyr10 and Tyr14 likely undergoing sulfation, while Tyr15’s sulfation is less evident. Additionally, the sulfated tyrosine residues Tyr100 and Tyr100c in antibody 412d interact with gp120. Ser7 (Serine 7), which contains an O-linked glycan, is a previously identified glycosylation site that may help stabilize the N-terminal configuration of CCR5. These extensive interfaces suggest a strong, high-affinity interaction between CCR5 and gp120 [21].
Further investigation is warranted to explore the physical linkage between CD4 and the coreceptor, given the potential synergistic effects that could enhance HIV-1 entry. Co-immunoprecipitation data indicates a robust association between CD4 and CCR5 on the cell surface, independent of Env. However, contradictory findings from immunomicroscopic studies raise questions about the extent of this association [92, 93]. In the CD4-gp120-CCR5 complex, the orientation of CD4 positions its TM domain considerably away from the TM helices of CCR5. Despite CD4’s TM domain bending back towards CCR5, direct contact between the TM domains of CD4 and CCR5 seems unlikely, mainly when both are bound to gp120. Further research is needed to elucidate the precise nature of the interaction between CD4 and the coreceptor, as well as its implications for HIV-1 entry.
HIV-1 entry into CD4+ T cells depends on the interaction between the Env and the coreceptors CXCR4/CCR5, which are determined by the virus’s tropism. This process begins when the Env glycoprotein gp120 binds to surface-expressed CD4, causing conformational changes [94] that enable gp120 to interact with coreceptors. This is a crucial step in initiating the fusion process (Fig. 2, Ref. [94], step 1). Following this, the gp41 subunit triggers fusion by embedding its hydrophobic fusion peptide into the lipid membrane after coreceptor binding and conformational alterations [95] (Fig. 2, step 2). The mechanisms of HIV-1 entry and viral membrane fusion have been thoroughly reviewed in other studies [21, 94, 96, 97]. The virus’s attachment to the plasma membrane is modulated by host proteins, such as P-selectin glycoprotein ligand-1 (PSGL-1) and CD43, which play a role in influencing HIV attachment [98]. HIV-1-encoded Vpu and co-clustered Gag proteins facilitate the target cell membrane attachment by downregulating PSGL-1 expression. Furthermore, interferon-induced TM proteins (IFITMs) impede HIV-1 entry by modulating fusion processes with the host membrane [99, 100].
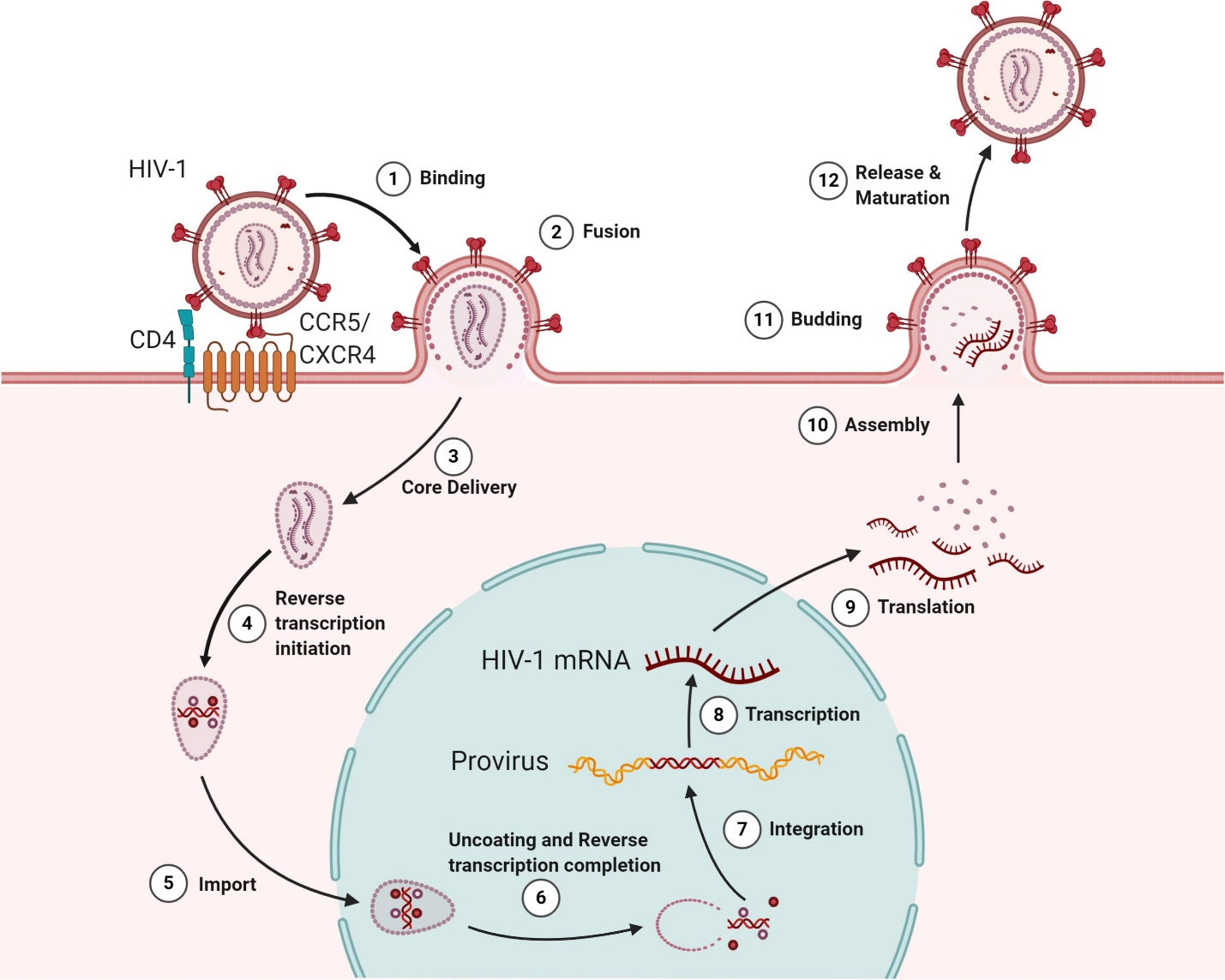
 Fig. 2.
Fig. 2.
The lifecycle of Human Immunodeficiency Virus (HIV). The infection initiates when the viral envelope glycoprotein binds to the CD4 receptor and coreceptors (CXCR4/CCR5) on the host cell surface (Step 1), facilitating viral entry and membrane fusion (Step 2). Once inside the host cell, the viral core is delivered (Step 3), and reverse transcription occurs in the cytoplasm (Step 4). The viral core is then transported into the nucleus (Step 5), where reverse transcription is completed, and the virus undergoes uncoating (Step 6). The enzyme integrase incorporates the viral genome into the host’s DNA (Step 7). Proviral transcription produces viral RNAs (Step 8), which are transported to the cytoplasm for protein synthesis (Step 9). Full-length viral RNA and proteins are assembled into new virions (Steps 10 and 11). Finally, the newly formed viral particles are released from the host cell, followed by maturation into fully infectious virions (Step 12). Reprinted (adapted) from [94] under a Creative Commons license. CXCR4, C-X-C chemokine receptor type 4.
Host factors such as IFITMs and SERINC5 can restrict the virus’s sensitivity to retroviral envelope glycoproteins [101, 102, 103]. IFITMs serve as critical innate immune restriction factors that inhibit viral membrane fusion, particularly in the context of HIV-1 and other enveloped viruses. These proteins exert their antiviral effects by modulating the biophysical properties of membranes at the sites of viral entry. IFITM3, for instance, induces negative membrane curvature via its amphipathic helix (AH), forming intraluminal vesicles (ILVs) and creating a mechanical barrier that prevents the formation of fusion pores [104]. Concurrently, IFITMs enhance membrane rigidity and lipid order, particularly in lipid-disordered domains, thereby increasing resistance to deformation during viral fusion. Their strategic localization in endolysosomal compartments and the plasma membrane, governed by posttranslational modifications, ensures the proximity-dependent restriction of viral entry [105]. Moreover, IFITMs directly antagonize the HIV-1 Env by impairing its processing, reducing its incorporation into virions, and promoting premature shedding of the surface unit (SU), which collectively diminishes viral infectivity. Additionally, IFITMs trap viral fusion at the hemifusion stage by creating an energetically unfavorable membrane environment, preventing fusion pore formation and promoting viral degradation. Beyond target cells, IFITMs can incorporate into nascent virions, reducing their infectivity independent of specific envelope glycoprotein interactions. Notably, IFITM2 and IFITM3 exhibit stronger antiviral activity than IFITM1, likely due to their predominant localization in the endosome. However, HIV-1 demonstrates adaptability through Env mutations that confer resistance to IFITM-mediated inhibition, highlighting a dynamic evolutionary interplay between host restriction factors and viral evasion strategies. This ongoing co-evolution has profound implications for HIV-1 pathogenesis and transmission, influencing viral fitness and informing potential therapeutic intervention strategies that target IFITM-virus interactions [106].
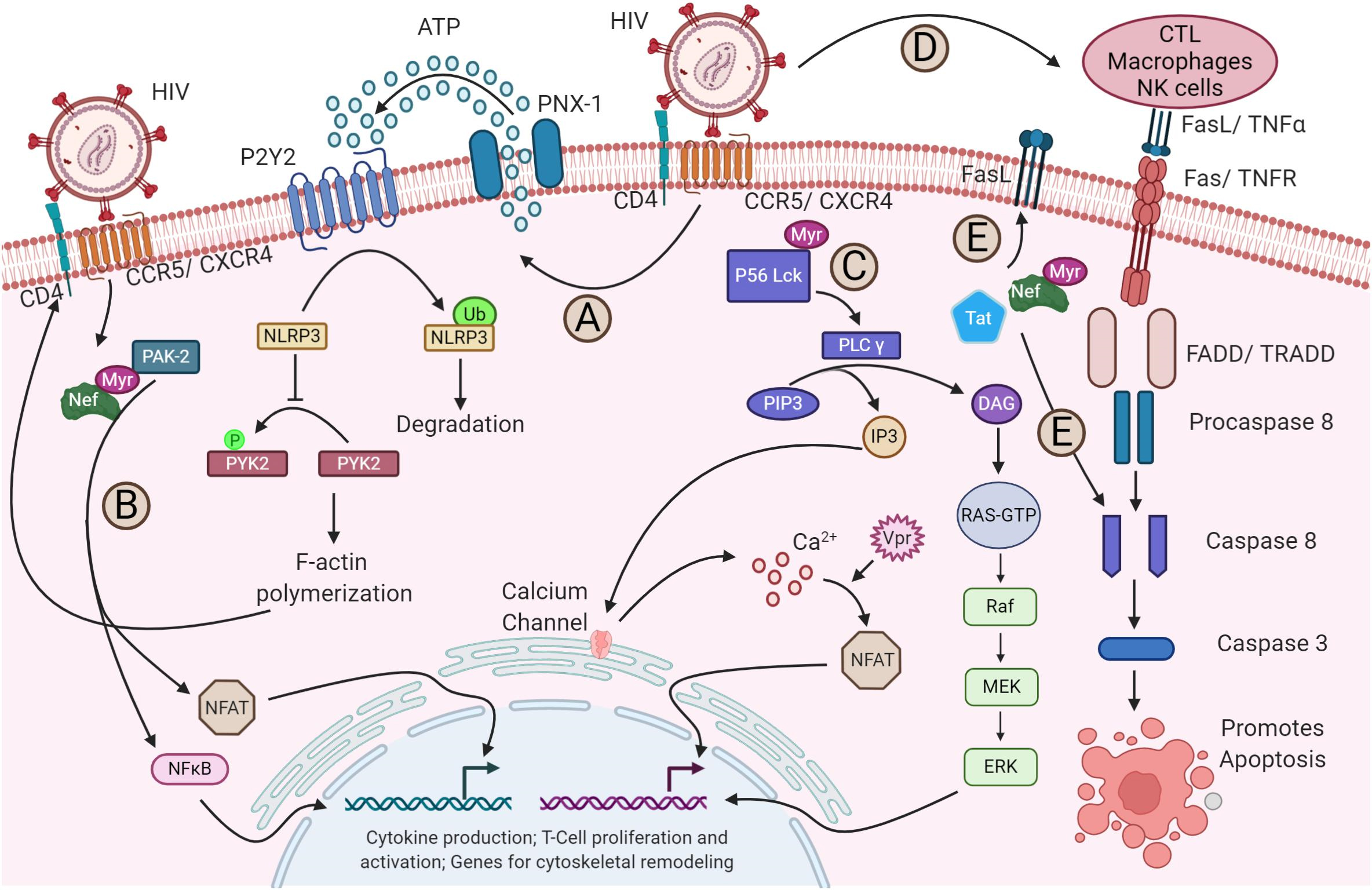
Upon HIV-1 binding to its receptors and coreceptors, various signaling pathways
are triggered and modified (Fig. 3, Ref. [94]). For instance, NACHT, LRR,
and PYD domains-containing protein 3 (NLRP3) inhibits F-actin remodeling, which
influences susceptibility to HIV-1 infection. The binding of the virus to its
receptors through P2Y purinoceptor 2 (P2Y2) signaling triggers the degradation of
NLRP3. Without NLRP3, proline-rich tyrosine kinase 2 (PYK2), a protein tyrosine
kinase, phosphorylates and activates cytoskeletal rearrangements, facilitating
the viral entry (Fig. 3A) [107]. Furthermore, the nuclear factor of activated T
cells (NFAT) and NF-
 Fig. 3.
Fig. 3.
HIV-1 modulation of host signaling pathways. (A) HIV-1 binding
to its receptor and coreceptor triggers P2Y2 activation by the release of ATP
through pannexin-1 (PNX-1), leading to the degradation of NLRP3. This, in turn,
enables PYK2 phosphorylation, which drives F-actin polymerization—a process
essential for viral fusion and entry. (B) The viral protein Nef activates nuclear
factor of activated T cells (NFAT) and NF-
In contrast, HIV-1 engagement with its receptor and coreceptors initiates the
activation of PLC-
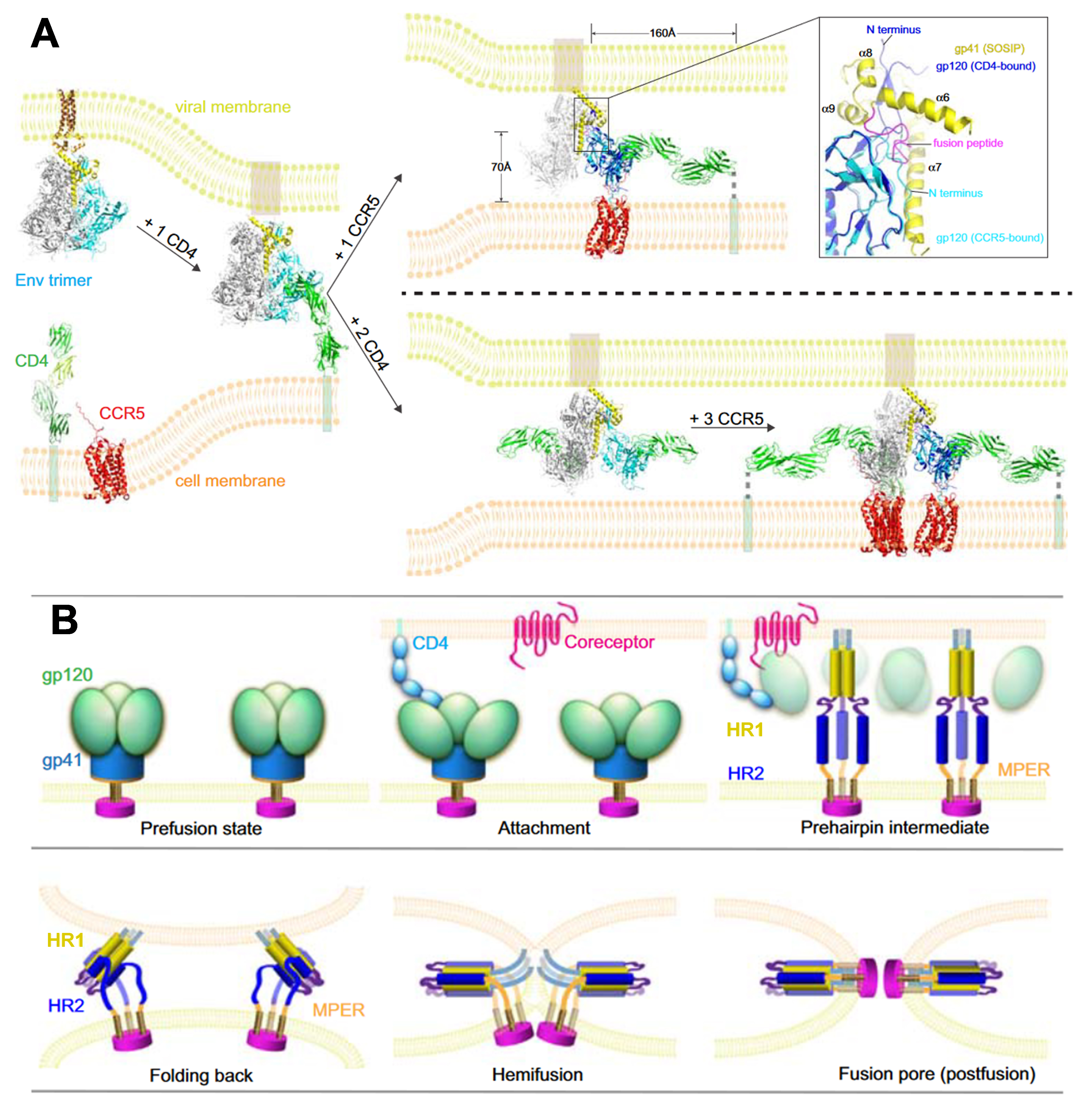
In structural biology, remarkable advancements have been made concerning HIV-1 Env and its interactions with cellular receptors. However, constructing a comprehensive molecular model presents challenges, chiefly due to the constraints of the latest structures. These limitations include artificial modifications applied to the Env constructs, which incorporate additional ligands to enhance stability, as well as the absence of membranes. Fig. 4 (Ref. [21]) illustrates a conceptual model depicting the initial stages of HIV-1 entry.
 Fig. 4.
Fig. 4.
HIV-1 entry protein interactions. (A) Molecular Model of
Initial HIV-1 Entry Steps: In its prefusion state, the unliganded Env trimer
ectodomain and MPER–TMD sit embedded within the viral membrane. Initial
attachment occurs through a single CD4 molecule binding to a single gp120 subunit
within the Env trimer. While the precise stoichiometry for Env, CD4, and
coreceptor interaction during fusion is not yet fully defined, CCR5 engagement
immediately after CD4 binding stabilizes Env’s conformational changes,
facilitating closer proximity to the host membrane. These shifts trigger the
outward extension of the fusion peptide, causing gp120 repositioning, potentially
leading to the dissociation of gp120 and the formation of the gp41 prehairpin
intermediate with its fusion peptide embedded in the target membrane. Key details
include a close-up of the gp120 N- and C-termini and gp41’s four helices
(
The initial attachment of HIV to the target cell surface begins with CD4 binding to the gp120 component of the Env protein. Although a single CD4 molecule can interact with the SOSIP trimer, this interaction does not trigger substantial structural changes, likely due to modifications like potently germline-targeting antibody 145 (PGT145) and donor strain (DS), which stabilize the trimer and prevent it from shifting into the open conformation typically induced by CD4 binding. Whether the Env trimer complex and single CD4 accurately represent a physiologically relevant transitional state is unclear. A single CD4 can induce conformational changes in Env, both in the presence and absence of PGT145 and with a DS mutation. Despite single-molecule (sm) FRET data suggesting that a single CD4 can elicit substantial changes, evidence from native, untriggered conformations of the prefusion SOSIP trimer disputes this [77]. The understanding of CD4-mediated attachment remains uncertain.
Additionally, the effectiveness of CD4-specific antibodies, such as ibalizumab, which target an epitope near the D1–D2 junction on CD4 opposite the gp120 binding site in inhibiting HIV-1 entry has yet to be conclusively proven [59, 113]. Recent studies employing advanced imaging techniques, including spectroscopy and microscopy, have revealed intriguing stoichiometry during membrane fusion among the Env trimer, CD4, and a coreceptor, possibly involving the oligomerization of both receptors [114, 115]. Previous studies indicate that the Env trimer remains functional even if one or more gp120 protomers cannot bind CD4 or the coreceptor, as a single CD4/coreceptor interaction is enough to initiate HIV-1 infection. Given that there are only approximately 14 Env spikes per virion, it is unlikely that an Env trimer engages three CD4s and three coreceptors simultaneously [19, 77, 116, 117].
A single CD4 molecule can induce conformational shifts in the Env trimer, promoting the formation of the coreceptor binding site. However, the exact role of the coreceptor in guiding further structural changes in gp120, which influence gp41’s fusion potential, remains under investigation. Notably, the recent structure of the CD4-gp120-CCR5 complex shows ambiguous allosteric changes in gp120 that might impact gp41. Comparative studies between gp120 bound to both CD4 and CCR5 versus gp120 bound solely to CD4 show no significant differences in gp120’s core [84, 118]. The main structural adjustments involve repositioning the V3 loop and reorientating gp120’s N- and C-termini near the gp41 interface.
Current understanding suggests that upon dissociation of the HIV-1 Env trimer
from gp120, the prefusion state of gp41 [74, 119, 120, 121, 122], which forms the “4-helix
collar”, undergoes a refolding process involving its four helices
(
In the absence of CCR5 binding, the necessity of a coreceptor, despite gp120 dissociation, to activate gp41 independently of CCR5 binding raises a crucial question. Primarily, gp120 dissociation without a coreceptor might prove nonproductive due to the spatial challenge. When the virion binds to the target cell surface with the Env trimer and interacts exclusively with CD4, the gp41 fusion peptide is approximately 160 Å away from the cell membrane surface (Fig. 4A). Premature dissociation of gp120 would prevent the fusion peptide from reaching the target membrane [120]. gp120 must bind to CCR5 to bridge this gap and facilitate membrane fusion, bringing the fusion peptide within a range of approximately 70 Å from the target membrane (Fig. 4A). This distance aligns with the proposed traversal distance of the fusion peptide during gp41 refolding to reach the target receptor [123, 124]. Consequently, CCR5 serves to promote membrane fusion by stabilizing CD4-induced conformational changes. Moreover, the formation of fusion pores likely necessitates the involvement of multiple Env trimers, as demonstrated in other viral fusion proteins [125]. A stable Env-receptor complex would be advantageous for consistent conformational changes across several Env trimers. Thus, to facilitate productive membrane fusion, a coreceptor stabilizes and anchors the conformation of the Env trimer induced by CD4. Therefore, even from a mechanical standpoint, the designation of the HIV-1 primary receptor and coreceptor appears serendipitous.
Theoretically, a single Env trimer can participate with three CD4 molecules and three CCR5 coreceptors, collectively facilitating trimer activation. This configuration would necessitate binding three CD4s and three coreceptors to a single Env trimer, bringing it close to the cell membrane and enabling productive fusion. HIV-1 employs alternative entry mechanisms that facilitate CD4-independent infection, significantly impacting viral persistence and reservoir establishment [126]. These pathways involve structural adaptations in the viral Env protein and the exploitation of host receptors, such as DC-SIGN, enabling viral dissemination while evading immune detection. Structurally, CD4-independent HIV-1 variants exhibit an enhanced intrinsic reactivity of Env trimers, requiring lower activation energy to transition between conformational states. This allows direct exposure of the coreceptor-binding site (e.g., CCR5/CXCR4) without CD4 priming, thereby facilitating premature gp41 coiled-coil formation and bypassing CD4-induced activation steps. However, this structural flexibility also increases susceptibility to neutralization due to the exposure of vulnerable epitopes, rendering such variants rare in vivo but highlighting HIV-1’s adaptability in selecting entry routes [127]. Additionally, dendritic cells contribute to viral persistence through the C-type lectin receptor DC-SIGN, which mediates HIV-1 capture and transmission via two distinct mechanisms. First, DC-SIGN binds to gp120 through carbohydrate-independent interactions, allowing virions to remain infectious within endosomal compartments for prolonged periods [128, 129].
Second, X4-tropic HIV undergoes low-level replication in DC-SIGN+ cells, generating new viral progeny for delayed transmission [124]. This enables dendritic cells to serve as long-term viral reservoirs, with subsequent trans-infection of CD4+ T cells occurring via synaptic transfer of intact virions or delivery of replication-competent viruses from intracellular compartments [129]. Furthermore, CD4-bypass mechanisms contribute to the formation of a latent reservoir by enabling HIV-1 entry into resting memory T cells, including CD4low central memory (TCM) and transitional memory (TTM) subsets, through DC-SIGN-mediated transfer [130]. These pathways facilitate immune evasion by reducing immune activation via endosomal entry routes that circumvent pathogen recognition receptors [131]. Additionally, latently infected memory T cells persist through IL-7-driven homeostatic proliferation and antigen-independent survival mechanisms [130]. Recent studies indicate that DC-SIGN+ dendritic cells in mucosal tissues preferentially transfer HIV-1 to long-lived memory T cells, thereby establishing stable viral reservoirs that are resistant to antiretroviral therapy [132, 133]. This alternative transmission route may underlie the rapid reseeding of viral reservoirs following treatment interruption, emphasizing the need for targeted therapeutic strategies to disrupt CD4-independent viral dissemination.
Moreover, the configuration of the CD4 molecule within the CCR5 complex, which aligns almost parallel to the TM helices of CCR5 and potentially parallel to the bilayer plane, may have implications for membrane function. If the CD4 ectodomain exhibits sufficient rigidity, the CD4-gp120-CCR5 complex formation could bring a local bend in the membrane. Investigating whether this potential membrane bending phenomenon can account for the observed blockage of CD4 and CCR5 colocalization on cell surfaces induced by gp120 when cholesterol depletion occurs, a condition known to promote membrane curvature, would be essential [134]. Additionally, further research is warranted to explore the potential roles of phospholipids during HIV-1 entry.
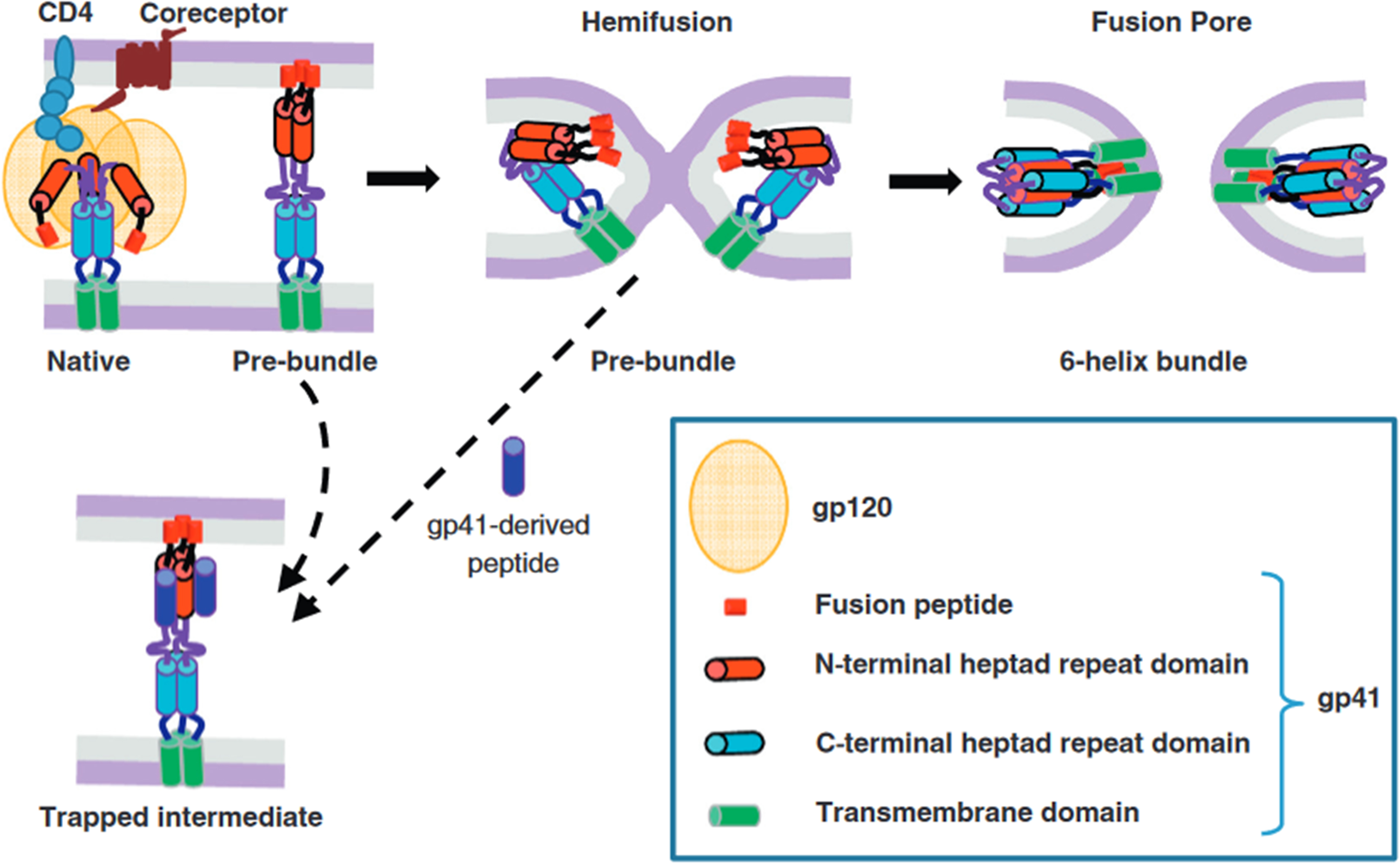
The fusion of two bilayer membranes is thermodynamically favorable, yet it encounters a formidable kinetic barrier [135, 136]. Fusion agents, including viral fusion proteins, effectively lower this barrier by harnessing the released free energy during the conformational changes of proteins (Fig. 4B, Fig. 5 (Ref. [137])). The merging of two bilayers into a single monolayer typically follows a general pathway, likely culminating in an intermediate state known as ‘hemifusion’, wherein the proximal leaflets of both bilayers have fused while their distal ones remain separate. This hemifusion intermediate is thought to possess a stalk-like structure (Fig. 4B), and in studies of viral protein-mediated fusion, it appears to play a crucial intermediate role [135, 138]. The kinetic barriers associated with forming and transitioning from this intermediate state are likely considerable.
 Fig. 5.
Fig. 5.
HIV Env-mediated membrane fusion and its inhibition. The key steps of HIV membrane fusion follow the engagement of the CD4 receptor and coreceptor by the gp120 subunit of the Env glycoprotein. The gp41 subunit undergoes a structural transition from its native conformation to various intermediate states, forming a stable six-helix bundle in the post-fusion state. This transformation involves the exposure of heptad repeat (HR) domains, depicted as red (N-terminal HR) and blue (C-terminal HR) cylinders. Inhibitory peptides derived from the C-terminal HR region (dark blue) can bind to the N-terminal HR (red), blocking the formation of the six-helix bundle and halting fusion. The dashed arrow between the hemifusion state and the peptide-bound intermediate indicates that even late-stage intermediates, including nascent fusion pores, can revert in the presence of these peptides, preventing complete fusion. Reproduced with permission from Gregory B Melikyan, HIV entry: a game of hide-and-fuse; published by Elsevier, 2014 [137].
The evidence accumulated to date strongly suggests that viral fusion proteins facilitate the reduction of the kinetic barriers inherent in membrane fusion. More significantly, they act as catalysts, driving and facilitating the process of membrane fusion, as follows:
Step 1: The initial stage involves the formation of protein bridges between bilayers as the fusion proteins undergo conformational changes (Figs. 4B,5). Across all viral fusion proteins examined thus far, two essential membrane-interacting elements are present: a TM anchor at the C-terminus, which secures the protein within the viral membrane, and a hydrophobic patch that binds to the target membrane. Moreover, these proteins typically adopt a trimeric configuration in their fusion-active state. The fusion reaction commences when the fusion protein binds to a ligand, often protons, in response to low pH in an endosome [96, 139]. Still, cellular or viral proteins sometimes trigger a conformational change that propels each subunit toward contact with the target membrane (Figs. 4B,5). Many fusion proteins are fragments of larger precursors, such as hemagglutinin subunit 2 (HA2) from the influenza virus hemagglutinin or gp41 from the HIV envelope protein. They must shed their N-terminal fragments to initiate the fusion process, which typically contain receptor-binding domains (e.g., HA1 or gp120 in the influenza virus hemagglutinin). While evidence for an extended intermediate is compelling, it remains indirect. The subsequent step involves collapsing into a folded-back conformation, often referred to as a ‘pre-hairpin intermediate’. Depending on the intermediate, the half-life could range from several minutes for HIV-1 gp41 to just a few seconds in other cases. Step 2: Following the bridge’s collapse, the fusion peptide or loop in the target membrane converges with the TM anchor (Figs. 4B,5). This collapse distorts the two bilayers, forming a nipple-like structure around a relatively confined area [140]. The fusion peptide, which perturbs the bilayer and lowers the distortion energy, may further potentiate this membrane distortion. Step 3: The distortion of the individual membranes reduces the energy barrier between separated and hemifused bilayers (not necessarily symmetrically, due to differing anchoring of the fusion protein at the two ends), leading to the formation of a hemifusion stalk between them (Figs. 4B,5). Step 4: A transient fusion pore is generated when the hemifusion stalk opens. During protein refolding, a final conformational change renders the open state irreversible (Figs. 4B,5). While some fusion proteins cause the pores to flicker open and close, others do not. Flickering may depend on the rapidity with which the conformational change is completed. At least two fusion-protein trimers are likely required to execute steps 3 and 4. Nonetheless, the precise number of participating trimers and their interaction dynamics remain topics of ongoing discussion [96, 139].
Central to this process is the formation of a stable six-helix bundle within the gp41 subunit, which provides the necessary free energy to drive membrane fusion. Structurally, the Env complex consists of the receptor-binding gp120 and the fusion-mediating gp41 subunits. The latter contains two functionally significant heptad repeat regions—HR1 (proximal to the N-terminal fusion peptide) and HR2 (adjacent to the transmembrane domain)—which undergo substantial rearrangements during membrane fusion [141]. In its native state, gp41 exists in a metastable prefusion conformation within the Env trimer. Upon CD4 receptor binding and subsequent coreceptor engagement, gp41 transitions through a series of intermediate states before forming the thermodynamically stable six-helix bundle, which juxtaposes viral and host membranes, facilitating fusion [142]. The postfusion six-helix bundle consists of a central trimeric HR1 coiled coil, into which HR2 segments pack in an antiparallel orientation, generating a trimer-of-hairpins configuration essential for overcoming energetic barriers to fusion [141, 142]. Structural studies have identified key intermediate states, including an asymmetric fusion-intermediate conformation stabilized by a MPER-specific neutralizing antibody, suggesting potential sites for therapeutic intervention [143, 144]. Molecular dynamics simulations indicate that the final postfusion conformation is stabilized by hydrophobic interactions between fusion peptides and transmembrane domains, forming an energetically favorable core [143]. The transition to the six-helix bundle is an exergonic process that releases sufficient free energy (estimated at 40–60 kT) to overcome kinetic constraints associated with membrane fusion [142, 143]. Experimental findings reveal that bundle formation is directly coupled to membrane merger, as evidenced by studies employing lysophosphatidylcholine (LPC) to inhibit hemifusion. LPC-mediated arrest of membrane fusion prevented six-helix bundle formation, an effect reversed upon LPC removal, confirming that the free energy released during the transition is a primary driver of pore formation rather than the bundle itself [142, 145]. These insights into gp41 structural transitions have profound implications for HIV therapeutics, particularly in the development of fusion inhibitors and vaccines. Notably, MPER-targeting broadly neutralizing antibodies can stabilize late-stage fusion intermediates, highlighting the potential for interventions that disrupt membrane fusion even after initiation [143].
To exemplify these generalizations, we consider the fusion proteins of three viruses, each with established 3D structures for pre-fusion and post-fusion states: influenza, dengue, and vesicular stomatitis. While traditionally classified as class I, class II, and class III viral fusion proteins [146, 147]. This classification can now be somewhat ambiguous and may obscure more than it elucidates; thus, it’s advisable to avoid it. Viral fusion proteins from paramyxoviruses and alphaviruses also exhibit structures in both conformational states [148, 149, 150, 151].
The initially metastable virus fusion protein must undergo a specific triggering event to initiate the fusion process (Figs. 4B,5). Different viruses employ various triggers, each tailored to the virus’s particular requirements. Current research identifies several distinct triggers and their corresponding viruses: receptor binding to a separate attachment protein (paramyxoviruses), receptor binding to fusion proteins (retroviruses), receptor plus coreceptor binding (HIV), receptor plus low pH (avian virus), receptor plus low pH (alphaviruses, flaviviruses, and influenza viruses), and receptor binding plus unknown triggers (Ebola). Despite the diversity of these trigger mechanisms, they all ultimately result in the refolding of the fusion protein into its stable conformation post-fusion—a process that is thermodynamically favored. Additionally, it has been observed that certain fusion proteins, such as influenza hemagglutinin, can be triggered for refolding and fusion by heating the virus [152, 153].
Upon triggering, many fusion proteins change oligomeric association as an initial response. For instance, the rhabdovirus G trimer [154] or flavivirus E protein homodimer [155] may undergo dissociation and rearrangement of interactions between fusion proteins. This process facilitates the reorientation and release of hydrophobic fusion peptides toward the target membrane by altering interunit interactions and inducing conformational changes within the fusion protein. Subsequently, after inserting the fusion peptide into the outer leaflet of the target membrane, it is transferred to the inner leaflet. In some cases, a viral fusion protein is initially inserted via a trimer, while in others, monomers are inserted as subunits [156], which are then induced by membrane interactions to orient and concentrate locally, thereby promoting trimerization. In addition to monomers, other subunits are inserted, which are then induced by membrane interactions that provide orientation and local concentrations, thereby promoting trimerization [97]. Consequently, a trimeric intermediate, sometimes referred to as a pre-hairpin, is formed, bridging the membranes of the target and viral infection.
In some fusion proteins, the timing of trimerization is unclear, and the extended intermediate remains poorly characterized. During this process, an external layer, linked to the viral membrane, surrounds the central trimer, which is embedded in the target membrane. Eventually, the structure transitions into a hairpin-like conformation, bringing the target membrane and fusion peptides closer to the viral membrane and its TM regions. In addition to these structural changes, the fusion process involves further intricate dynamics. Notably, lipidic stalks or hemifusion intermediates emerge due to the fold-back process (Figs. 4B,5) [157]. Within these intermediates, a transient, small fusion pore initially opens, representing a critical juncture in the fusion cascade [158, 159]. Disruptions of viral membranes can arise from hairpin formation-induced stresses and interactions of the fusion protein’s membrane-proximal regions [52, 139, 160]. As a result of the fold-back process (Figs. 4B,5), lipidic stalks or hemifusion intermediates are formed [135]. This pore subsequently expands irreversibly, facilitating the crucial exchange of genetic material and infection between the virus interior and the host cell cytoplasm [136, 161]. Interestingly, the poring process, which involves multiple fusion proteins [139, 162], is recognized as the most energetically demanding phase of the fusion process. Despite the diverse architectural arrangements of pre-fusion states among different viral fusion proteins, they all ultimately adopt trimeric hairpin structures in their post-fusion forms.
The complexities surrounding HIV-1 entry present an intricate and challenging puzzle in molecular virology, with profound implications for vaccine development and therapeutic innovation. A thorough understanding of this process not only advances HIV-1 treatment but also provides insights into membrane fusion mechanisms shared across multiple enveloped viruses. Despite significant progress, key molecular aspects of HIV-1 entry remain unresolved, necessitating further investigation into full-length Env in its native membrane environment to elucidate its conformational changes and interactions with cellular receptors and HIV-1 matrix proteins. Advances in cryo-EM and live-cell imaging techniques offer promising avenues for real-time molecular visualization, potentially replacing theoretical models and deepening our understanding of viral fusion dynamics. Future research should integrate structural biology, immunotherapy, and gene-editing strategies to overcome the current limitations of therapeutic approaches. Next-generation approaches should combine multi-epitope bnAbs with small-molecule inhibitors to counteract viral escape, leveraging structural insights into the dynamics of the Env trimer. Host-directed therapies, such as pharmacological upregulation of restriction factors and clustered regularly interspaced short palindromic repeats (CRISPR)-based interventions, hold potential for enhancing innate antiviral defenses. Cure-focused strategies should explore gene editing (e.g., CCR5 knockout) in combination with latency-reversal agents and long-acting fusion inhibitors to eliminate viral reservoirs. Additionally, computational approaches can accelerate the design of bispecific bnAbs and broad-spectrum entry inhibitors, facilitating clinical translation for HIV prevention and cure. By integrating these interdisciplinary efforts with cutting-edge imaging and computational techniques, future research will not only address persistent challenges in HIV-1 entry inhibition but also uncover fundamental principles of membrane fusion relevant to multiple viral families.
TM, Transmembrane; AIDS, Acquired Immunodeficiency Syndrome; BS, Binding Site; CCR5, C-C Chemokine Receptor 5; CCL5/RANTES, C-C Motif Chemokine Ligand 5/Regulated on Activation, Normal T Cell Expressed and Secreted; CD4, Cluster of Differentiation 4; CD43, Cluster of Differentiation 43; CRS2, Conserved Residue Site 2; CXCL12/SDF-1, C-X-C Motif Chemokine 12/Stromal Cell-Derived Factor 1; CXCR4, C-X-C Chemokine Receptor 4; cryo-EM, Cryo-Electron Microscopy; cryo-ET, Cryo-Electron Tomography; DAG, Diacylglycerol; ECL2, Extracellular Loop 2; ECL3, Extracellular Loop 3; EM, Electron Microscopy; Env, Envelope Glycoprotein Complex; ER, Endoplasmic Reticulum; ERK, Extracellular Signal-Regulated Kinase; Fab, Fragment Antigen-Binding; FRET, Förster Resonance Energy Transfer; Gag, Group-Specific Antigen; gp120, Glycoprotein 120; gp41, Glycoprotein 41; HA, Hemagglutinin; HA1, Hemagglutinin Subunit 1; HA2, Hemagglutinin Subunit 2; HIV, Human Immunodeficiency Virus; HIV-1, Human Immunodeficiency Virus Type 1; HR, Heptad Repeat; HR1/HR2, Heptad Repeat 1/Heptad Repeat 2; IFITMs, Interferon-Induced Transmembrane Proteins; IFN, Interferon; IP3, Inositol 1,4,5-Trisphosphate; Lck, Lymphocyte-Specific Protein Tyrosine Kinase; LLP, Lentiviral Lytic Peptide; MAPK, Mitogen-Activated Protein Kinase; MHC, Major Histocompatibility Complex; MIP-1
AE conceptualized the study and wrote the original draft. HE and HM: designed the study, collected the data, created the figures, and reviewed the study. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work. All authors read and approved the final manuscript. All authors contributed to editorial changes in the manuscript.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
During the preparation of this work the authors used ChatGPT in order to check spell and grammar of whole the document. After using this tool, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.