







 , Vanessa Pitozzi 2, Silvia Pontis 2, Paola L. Caruso 2, Sofia Beghi 2, Mariafrancesca Caputi 3, Marcello Trevisani 2, Francesca Ruscitti 2,*
, Vanessa Pitozzi 2, Silvia Pontis 2, Paola L. Caruso 2, Sofia Beghi 2, Mariafrancesca Caputi 3, Marcello Trevisani 2, Francesca Ruscitti 2,*1 Department of Neuroscience, Psychology, Drug Area and Child Health, University of Florence, 50139 Florence, Italy
2 Target Innovation Department, Global Research and Preclinical Development, Chiesi Farmaceutici S.p.A., 43122 Parma, Italy
3 Department of Medicine and Surgery, University of Parma, 43126 Parma, Italy
Abstract
Background: Idiopathic pulmonary fibrosis (IPF) is a chronic and
progressive interstitial lung disease (ILD) whose cause and pathogenesis are not
yet well understood. Until now, no animal model of lung fibrosis succeeds in
recapitulating all IPF features, thus the use of different rodent models is
essential for the evaluation and development of new effective pharmacological
treatments. Recently, the alveolar epithelial dysfunction has been emphasized in
the etiopathogenesis context of IPF. Remarkably, the role of an aberrant basaloid
cell type, primarily found in humans and confirmed in mice, seems to be crucial
in the establishment and progression of the disease/model. Our work aimed to
characterize for the first time this cell population in a rat model of lung
fibrosis induced by a double bleomycin (BLM) administration, demonstrating the
translational value of the model and its potential use in the testing of
effective new drugs. Methods: Rats received an intratracheal BLM
administration at day 0 and 4. Animals were sacrificed 21 and 28 days post-BLM.
The fibrosis evaluation was carried out through histological (Ashcroft score and
automatic image analysis) and immunoenzymatic analysis. Immunofluorescence was
used for the characterization of the aberrant basaloid cells markers. Results: Lung histology revealed an increase in severe grades of
Ashcroft scores and areas of fibrosis, resulting in a rise of collagen deposition
at both the analyzed time-points. Immunofluorescence staining indicated the
presence of KRT8+ cells in bronchial epithelial cells from both controls (saline,
SAL) and BLM-treated animals. Interesting, KRT8+ cells were found exclusively in
the fibrotic parenchyma (confirmed by the alpha-smooth muscle actin
(
Keywords
- idiopathic pulmonary fibrosis
- IPF
- aberrant basaloid cells
- fibroblastic foci
- progressive pulmonary fibrosis
- KRT8+ epithelial cells
- bronchiolization
- bleomycin
Idiopathic pulmonary fibrosis (IPF) is the most common Interstitial Lung Disease (ILD), defined by the American Thoracic Society (ATS) as “a specific form of chronic, progressive, fibrosing interstitial pneumonia of unknown cause, occurring primarily in older adults, and limited to the lungs” [1]. Although the cause(s) of the disease is/are not yet understood, several risk factors are associated to it, such as environmental and occupational exposures, male sex, age and genetic factors [2]. IPF is characterized by collagen and extracellular matrix components deposition, clustered cystic airspaces called honeycomb areas with fibroblastic foci, increased airway wall thickness and apoptosis of alveolar epithelial cells. These features compromise the alveolar repair and function, leading to gas exchange decrease and pulmonary function decline, consequently patients have respiratory symptoms such as dyspnea and cough [2, 3, 4].
Until now, studies conducted on the pathogenesis of IPF suggest that lung fibroblast and epithelial cell activation, as well as the secretion of fibrotic and inflammatory mediators, have been strongly associated with the development and progression of IPF. Recent studies indicated that the deregulation of the alveolar epithelium is a crucial event for the development of the disease. Especially, repetitive micro-injuries to alveolar epithelial cells lead to abnormal extracellular matrix (ECM) accumulation and lung remodeling caused by a defective epithelial-fibroblast communication [3, 5, 6]. Airway-specific pathogenic features of IPF include bronchiolization of the distal airspace with the replacement of the distal alveolar type 1 and 2 (AT1 and AT2) epithelial cells with proximal lung cells, in particular basal cells [4, 7]. Two independent groups [8, 9] recently identified, by a single cell RNA sequencing approach, a unique population of cells in the lung of IPF patients, called “aberrant basaloid” or “KRT5-/KRT17+” cells that co-express markers of basal epithelial cells (KRT17 but not KRT5), mesenchymal markers (Vimentin), and senescence-related genes (CDKN1A). Especially, aberrant basaloid cells were found at the edge of myofibroblast foci in the distal lung parenchyma suggesting their role in the active tissue remodeling [4, 6, 7, 8, 9]. It was pointed out that AT2 cells have diminished capacity for transdifferentiation into AT1 cells in the IPF lungs, a phenomenon that could trigger an aberrant tissue repair process. This mechanism may induce AT2s to generate a permanently intermediate phenotypic state, identified as aberrant basaloid, which cause loss in AT1 cells in the lung [10, 11]. Based on these findings, Strunz et al. [12] and others [6, 13] demonstrated the presence of an intermediate cell population, endowed with a similar transcriptional profile to aberrant basaloid cells of IPF patients and named KRT8+ alveolar differentiation intermediate (ADI), in the lung of bleomycin (BLM)-treated mice. Moreover, the Authors confirmed the transition state nature of KRT8+ cells by demonstrating the co-expression of AT2 markers, such as Prosurfactant protein C (Pro-SPC). The BLM mouse model has become the most widely used and best-characterized animal model available for preclinical testing [14]. However, no data is available on the potential presence of KRT8 and Pro-SPC positive cells in the lung of rats receiving a double administration of BLM. The original reported data confirm the presence of KRT8+ cells into the lung of BLM-treated rats and support the use of the rat model, together with the mouse one, to further characterize the antifibrotic profile of candidate drugs in development for IPF.
Male Sprague Dawley rats (Charles River Italia, Lecco, Italy), weighing 200–250 g, were housed 3 or 2 per cage in standard conditions (light 7 a.m.–7 p.m., dark 7 p.m.–7 a.m., temperature 10 °C–24 °C, relative humidity 40%–70%) at arrival. Before any use, animals followed an acclimatization period of 5 days and no prophylactic or therapeutic treatment were administered to any animals during this time. Animals had free access to tap water and were fed with a standard certified laboratory mouse diet ad libitum (4RF21, Mucedola srl, Milan, Italy for Charles River Italia). They were checked for abnormalities or sign of health problem after arrival in the animal facility. All the procedures involving animals were reviewed, approved and authorized by the Italian Public Health System for Animal Health and Food Safety within the Italian Ministry of Health (MoH) (authorization number 246/2021-PR). All the procedures were performed within a certified animal facility: AAALAC (American Association for Accreditation of Laboratory Animal Care, https://www.aaalac.org/). Experiments were performed in full compliance with the European ethics standards in conformity with directive 2010/63/EU, Italian D. Lgs 26/2014, the revised ‘Guide for the Care and Use of Laboratory Animals’ (Guide for the Care and Use of Laboratory Animals, 1996), and the ARRIVE guidelines (Animal Research: Reporting of In Vivo Experiments).
Rats were randomized after weighing and body weight were checked every day before treatment to obtain an indication about health conditions. According to published protocol for this model [15], a total of 20 animals received intratracheal BLM (batch 100012570, Baxter Oncology GmbH, Halle, Germany) administrations at the starting point of the experiment and in the fourth day, using a dose of 1.5 U/kg/0.6 mL, as shown in Fig. 1. Especially, rats were anesthetized in cages through a sevoflurane gas (4% in oxygen) and then attached by incisor teeth to a support inclined about 45° respect to the work surface. Holding the mouth open, tongue was moved with tweezers and the trachea was observed using a small otoscope (000A3754, Welch-Allyn Li-Ion, Hallowell EMC, Pittsfield, MA, USA) accessorized of a rat intubation speculum (200A3588, Hallowell EMC, Pittsfield, MA, USA). While the vocal cords were opened, bleomycin was injected through a metal canula. The same procedure was carried out for the control group composed of 14 rats which received a 0.9% saline solution (batch 19TAAHC0, Sodium Chloride 0.9%, Eurospital S.p.A, Trieste, Italy), the same used for the BLM dilution. Animals were sacrificed at two different time points to investigate alveolar epithelial dysfunction at different stages of the disease: 6–8 rats for the control group at day 21 and day 28, respectively, and 9–11 BLM-treated rats at day 21 and 28, respectively. They were anesthetized with pentothal sodium solution (batch 23PT001A/1, MSD Animal Health, Rahway, NJ, USA) injected intraperitoneally and sacrificed by bleeding from the abdominal aorta. Lungs were washed through transcardiac perfusion with a saline solution and explanted. Left lobes were separated and used for histological analysis, while right lobes were allocated for markers quantification.
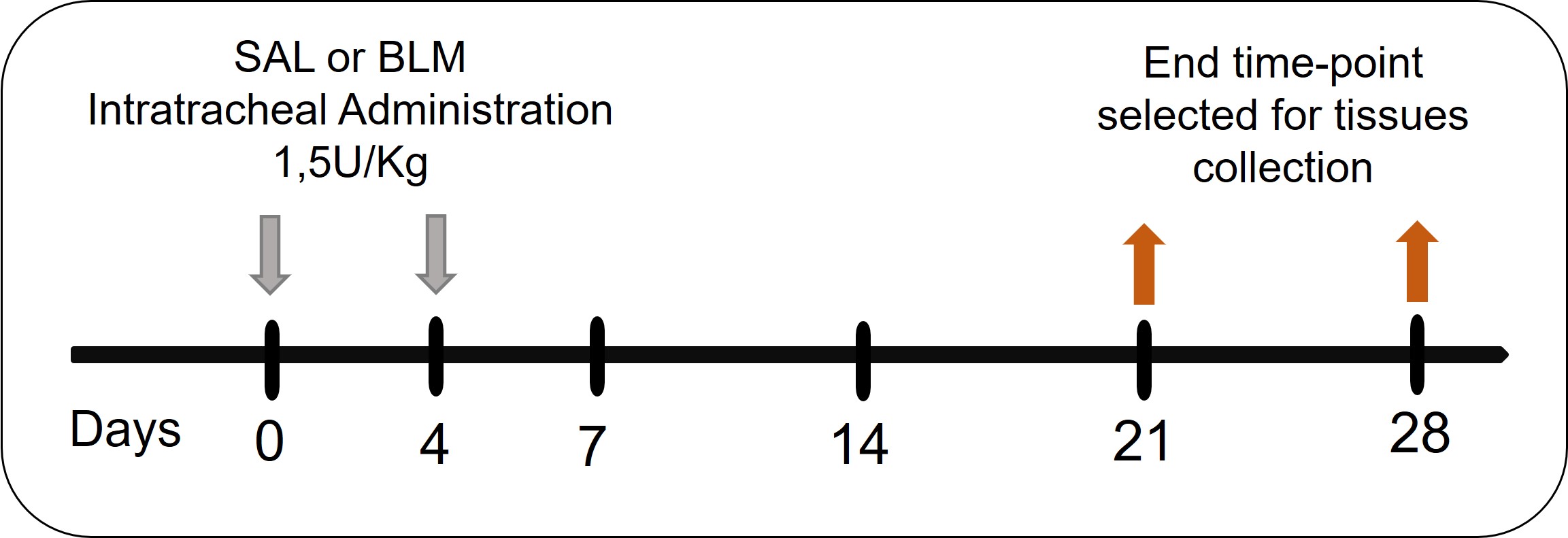 Fig. 1.
Fig. 1.
Experimental protocol. BLM, bleomycin; SAL, saline.
Left lobes were insufflated through a tracheal cannula with a 10% formalin
buffer, necessary to expand and fix the lungs and stored in test tubes filled
with 10% formalin buffer (Sigma-Aldrich, Saint Louis, MO, USA). Then, the formalin fixed lungs, were paraffin embedded
(FFPE). Each longitudinally oriented lung was cut to obtain 5 µm thick
sections representing the long axis of the entire parenchyma on slides. After
that, they were stained with the Masson Trichrome staining (activities performed
by Histolab Verona, Verona, Italy) that allowed to highlight collagen deposition
and fibrotic areas into the lung parenchyma. The slide images are acquired using
the NanoZoomer S-60 Digital scanner (Hamamatsu Photonics K.K., Shizuoka, Japan)
at 20
Immunofluorescence was carried out on lung sections of BLM and vehicle treated
animals using the Leica BOND RX automated stainer (BOND Research Detection System
DS9455, Leica Biosystems, Nussloch, Germany). Slides underwent an unmasking step
using EDTA buffer pH 8.5 and consequently they were incubated for 15 minutes with
the blocking solution composed of 10% of the donkey serum (D9663, Sigma-Aldrich,
Saint Louis, MO, USA) in PBS 1
The mean fluorescence intensity of KRT8, alpha-smooth muscle actin
(
Frozen right lobes were weighted and homogenized in 10 mL of ice-cold 1
The statistical analysis was performed using GraphPad Prism 10 software version
10.0.2 for Windows (GraphPad Software, San Diego, CA, USA). All data were
reported as mean
To assess the lung fibrosis induced by BLM, we analyzed representative Masson’s Trichrome-stained lung tissue sections comparing BLM-treated animal, at two different time points, and the relative controls (Fig. 2A). The staining highlighted severe fibrotic areas starting branching in rats receiving BLM at both day 21 and day 28. Moreover, large areas of normal lung parenchyma were replaced with dense collagen deposition as fibrotic single masses or confluent conglomerates corresponding to an Ashcroft score around 4 (colored in blue by Masson’s staining), while these features were absent in SAL rats. It is also possible to appreciate that BLM injury resulted in a patchy distribution of fibrotic lesions consisting of normal lung architecture in alternance to affected areas.
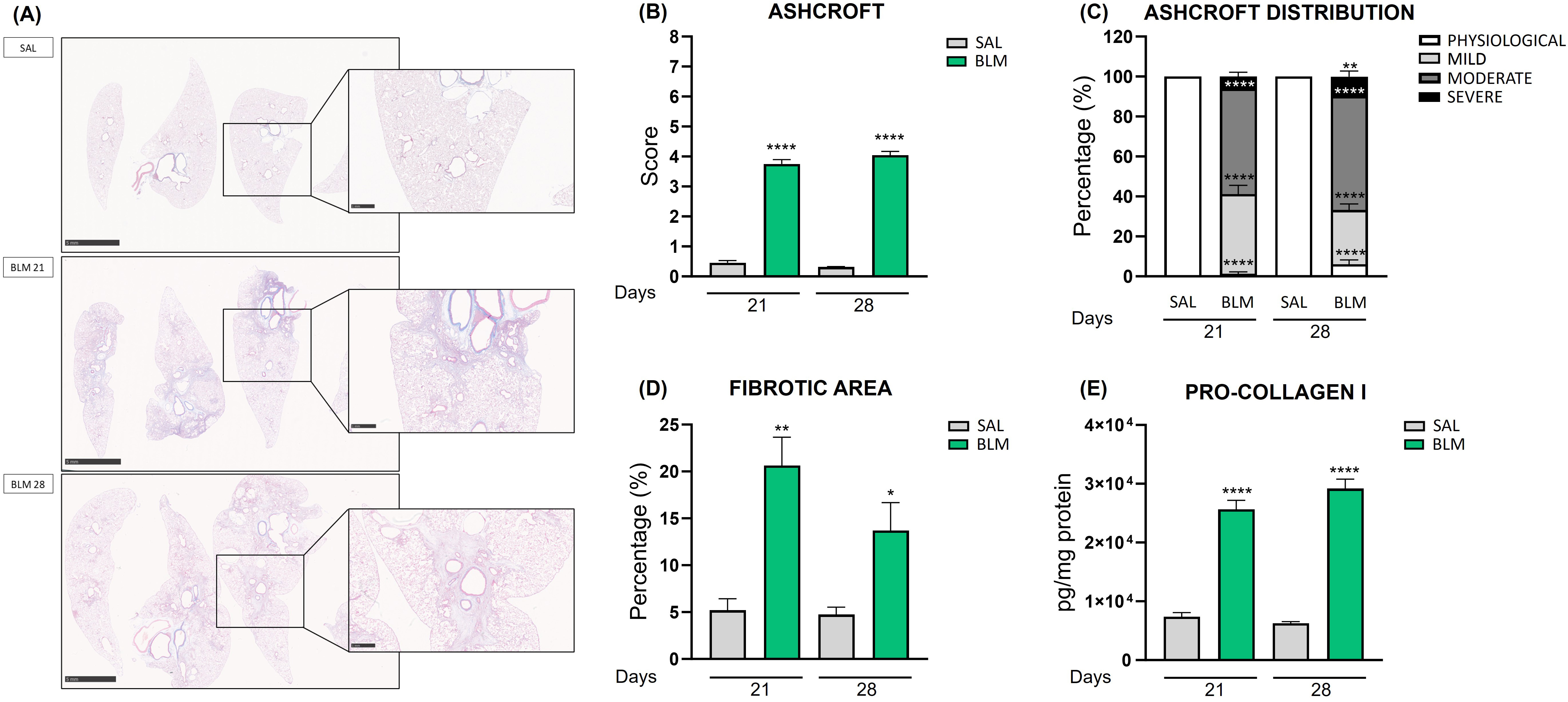 Fig. 2.
Fig. 2.
Quantification of fibrosis in rats treated with
bleomycin. (A) Masson’s Trichrome-stained lung tissue of the control group and
the BLM-treated animals at 21 and 28 days. Scale bar 5 mm or 1 mm. (B) The bar graph shows the Ashcroft
Score quantification of lung damage in different experimental groups treated with
BLM. ****p
The fibrotic condition was confirmed by Ashcroft score evaluation, using a
method based on the Ashcroft scale [16, 17]. BLM rats at 21 days reached a score
of 3.75 (p
Further confirmation of the fibrosis development in the rat BLM model was the
activation of fibroblasts into myofibroblasts. Notably, the immunofluorescence
staining of alpha-smooth muscle actin (
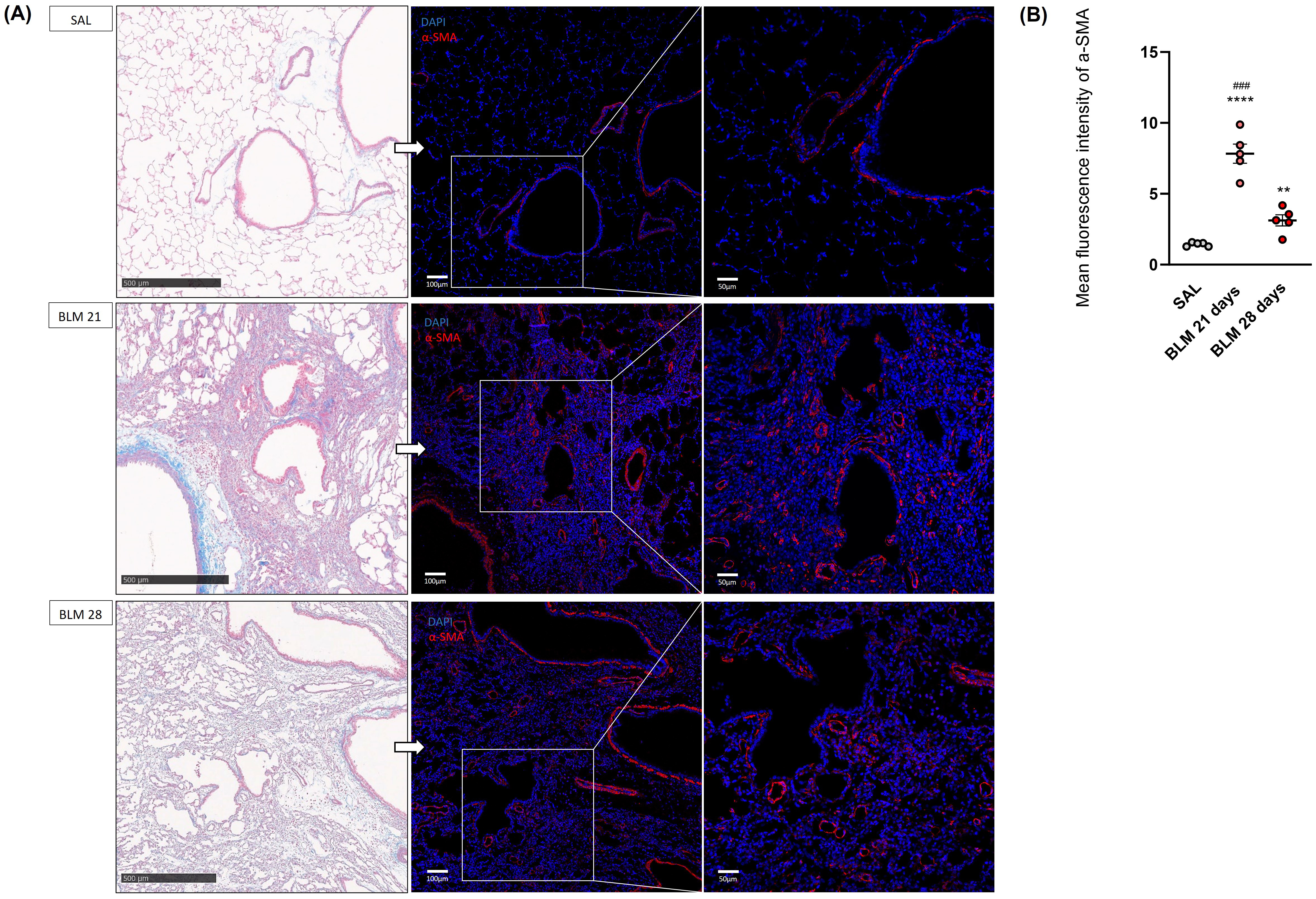 Fig. 3.
Fig. 3.
Histological and immunofluorescence evaluation of rat
lungs at 21 and 28 days after SAL or BLM treatment. (A) Representative images of
the parenchyma lung sections of rats receiving BLM or saline at 21 and 28 days.
On the left tissue lungs stained with Masson’s Trichrome, scale bar 500
µm. On the right, correspondent immunofluorescence staining of
myofibroblasts colored in red using the
The quantification of the
To further investigate the presence of aberrant basaloid cells in BLM-induced
fibrotic lesions, lung sections of BLM and SAL treated animals were stained with
the KRT8 and
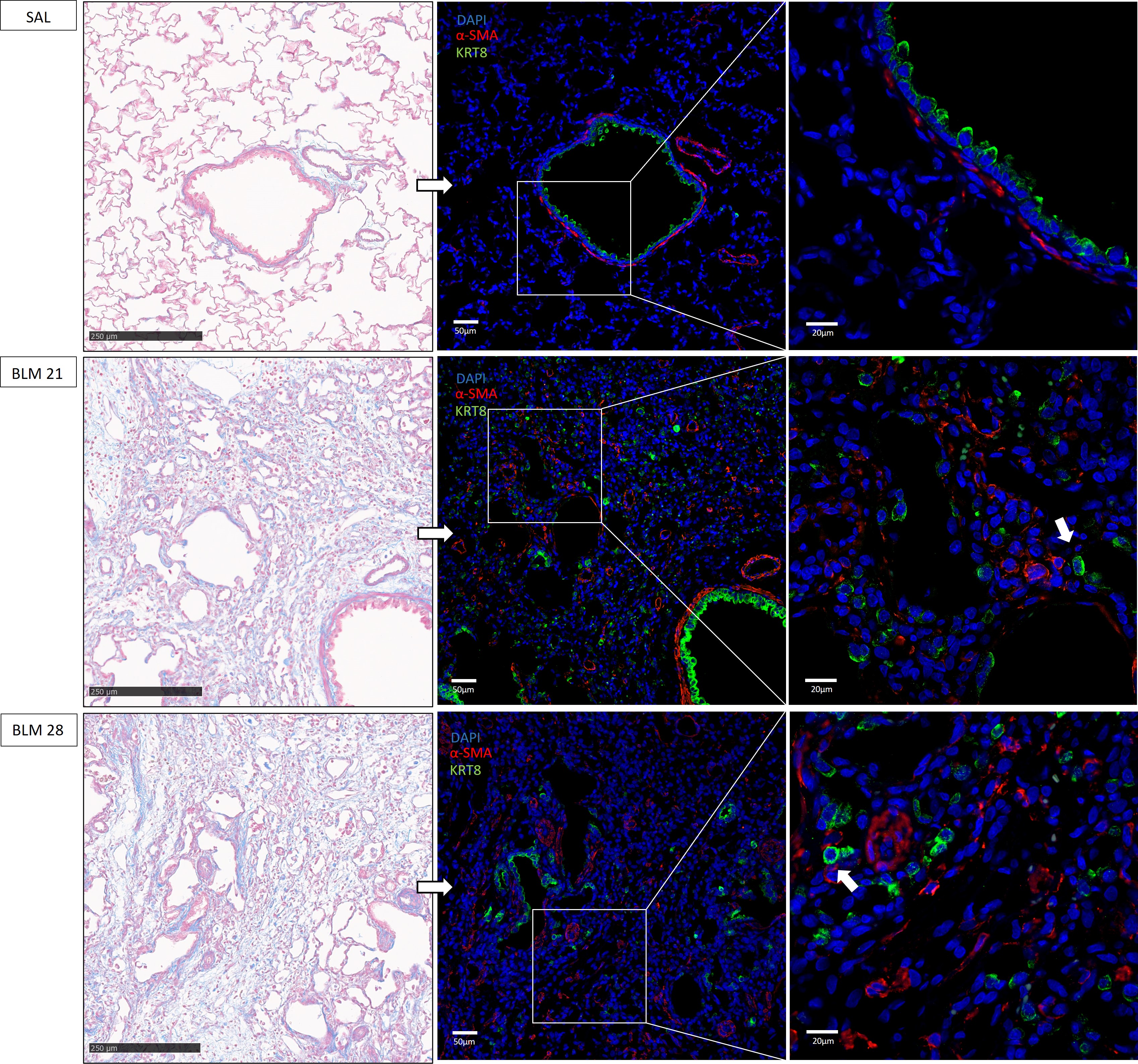 Fig. 4.
Fig. 4.
Representative immunofluorescence images of aberrant
basaloid cells in rat lungs. On the left tissue lungs stained with Masson’s
Trichrome, scale bar 250 µm. On the right, corresponding
immunofluorescence staining of aberrant basaloid cells colored in green using
KRT8 antibody,
It was hypothesized that aberrant basaloid cells originate from AT2 cells, acting as an intermediate state [12, 13, 18, 21, 22, 23], thereby preventing differentiation into AT1 cells. Immunofluorescence images obtained on our rat BLM model strengthened this assumption. Indeed, KRT8+ of rats on the twenty-first and twenty-eighth day showed a merged staining with the Pro-SPC, in contrast to controls, as shown in Fig. 5A (indicated with pink arrows). Furthermore, it was interesting to note how some aberrant cells expressed exclusively KRT8 (indicated with white arrows), definitely losing the marking of alveolar cells.
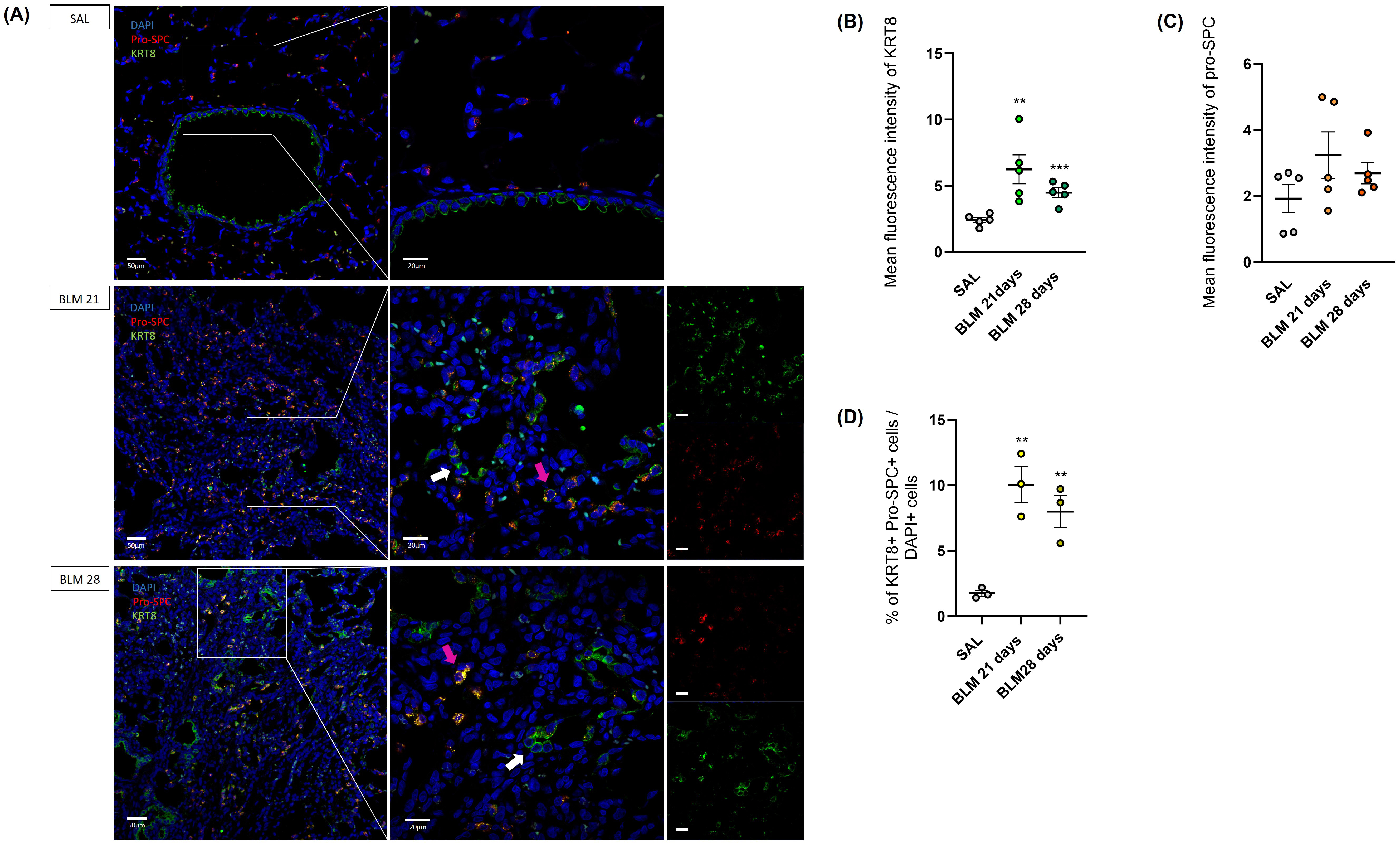 Fig. 5.
Fig. 5.
Immunofluorescence staining and quantification of alveolar type II (AT2)
and aberrant basaloid cells in rat lungs. (A) Immunofluorescence staining of
aberrant basaloid cells. KRT8 colored in green, pro-SPC in red and nuclei (DAPI)
in blue. Magnification 20
The highest presence of KRT8+ cells in the BLM-treated rats at 21 (p
The total number of transient AT2 cells were counted considering their
co-expression of the KRT8 and Pro-SPC markers: BLM rats at day 21 and day 28
exhibited a significantly higher proportion of KRT+ Pro-SPC+ cells compared to
controls (p
In addition, we evaluated the expression of Vimentin, a mesenchymal marker upregulated in EMT. As showed in Fig. 6 Vimentin was upregulated and localized in the lung parenchyma of BLM rats both at 21 and 28 days. According to other studies [4, 8, 9, 12, 21, 23, 25], where mesenchymal features of aberrant basaloid cells were described, we found colocalization of the immunofluorescence signal of KRT8 and Vimentin in both BLM rats at 21 and 28 days, depictured in Fig. 6 (indicated with white arrows). These data suggested the involvement of KRT8+ in the epithelial-mesenchymal transition process.
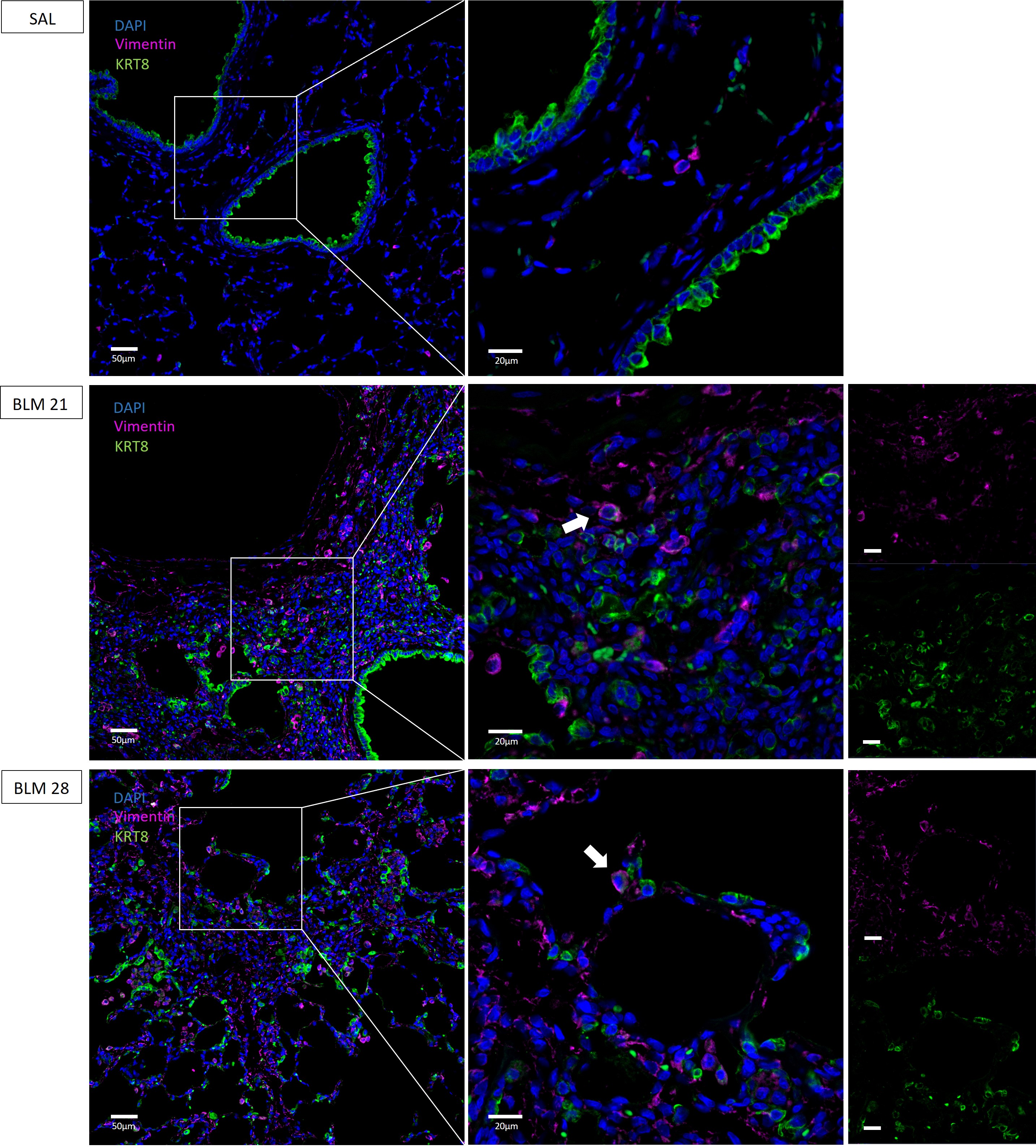 Fig. 6.
Fig. 6.
Immunofluorescence co-staining of KRT8+ cells and
Vimentin in rat lungs. KRT8 colored in green, Vimentin in purple and nuclei
(DAPI) in blue. Magnification 20
IPF is a high medical need rare and progressive lung condition that belongs to the heterogeneous group of diffuse interstitial lung diseases. The sole two approved drugs, Nintedanib and Pirfenidone, are unable to stop the disease progression and to cure and solve the patient’s clinical condition [1]. The lack of efficacious pharmacological therapies underlies the poor knowledge of the IPF pathogenesis and the need for relevant preclinical models having a high translational value. Efficacy of anti-inflammatory treatments such as steroids, is unproven. Etiopathogenetic mechanisms of fibrosis in IPF remain indefinable, with favored concepts of disease pathogenesis involving recurrent microinjuries to a genetically predisposed alveolar epithelium, followed by an aberrant reparative response characterized by excessive collagen deposition. Recently a new cell population named “aberrant basaloid cells” was identified in the lungs of IPF patients. This cell type seems to have an important role in epithelium dysfunctions and remodeling observed in IPF [9]. It has been hypothesized that aberrant basaloid cells activity may have important pathogenetic roles and that next-generation therapies may be directed toward this cell type in IPF and other fibrotic states in which they appear [26].
Recently, studies conducted by Strunz et al. [12], and Kobayashi et al. [27], demonstrated the presence of a cell population defined KRT8+ ADI in different mouse lung injury models, with a transcriptional profile similar to the KRT5-/KRT17+ cells identified in the IPF lung [23]. Even if the bleomycin model in mice is considered the first line animal model for preclinical testing, the Official American Thoracic Society guidelines recommended a confirmatory test on a second species in the phase of target validation and/or preclinical development to fully consolidate a preclinical proof-of-concept for drug candidates moving into the clinical development in IPF [13].
The current work had the aim to assess whether a KRT8+ ADI-like population of cells were present as well in a recently described rat model of lung fibrosis [15] induced by a double BLM intratracheal administration. The induction of fibrosis in our in-house model was supported by several endpoints including histological evaluations and biochemical measurements [14, 28]. Indeed, the Masson Trichrome-stained lung tissues showed collagen deposition and fibrotic areas exclusively in the BLM rats and the Ashcroft score evaluation confered a score of approximately 4 to BLM-treated animals at both day 21 and 28 compared to controls. The previous data was confirmed by an automatic image analysis software (VIS analysis Protocol Package), attributing a total percentage of fibrosis around 70% to animals receiving BLM, and lastly, the Pro-collagen 1 quantification in lung homogenates demonstrated the highest production of this protein in animals receiving the insult at both time points. As already described by our co-workers in a previous time course study [15], BLM-treated rats showed a peak of fibrosis at day 21 with a gradually decrease up to day 28 evaluated by histological analysis. On the contrary, in the BLM mouse model, as demonstrated by co-workers [29], fibrosis evaluated by micro-computed tomography (micro-CT) and histological analysis increased at day 7, peaking at day 14. Taken into consideration all the internal data validation on the two species, suggesting that rats and mice behave differently to BLM injury and show a different window of fibrosis onset and progression, we considered to analyse day 21 and 28 as the best terminal time points for the investigation of aberrant population.
The fibroblasts activation and their transition to myofibroblasts in BLM rats
were supported by the
Our results suggested the presence of a KRT8+ cell population in the lung of rats treated by a double administration of BLM. Especially, KRT8+ cells were observed through immunofluorescence at both investigated timepoints, 21 and 28 days after BLM administration. Stained KRT8+ cells appeared localized into the bronchi of both treated (BLM) and control rats, suggesting a common expression in the simple bronchial epithelium in the two conditions. Interestingly, they were selectively found in the fibrotic parenchyma and nearby myofibroblasts of BLM-treated animals and not SAL animals, as reported by previous studies on the mouse model [12, 13, 19]. Thus, these KRT8+ cell type that accumulates and persists up to 28 days post BLM-administration, could be responsible for the establishment of an aberrant tissue repair process and ECM deposition in the rat lung. Particularly, BLM rats on day 28 showed less KRT8+ cells compared to day 21, as shown by the fluorescence intensity analysis, further confirming the observed gradual reduction in fibrosis of the rat model, feature not found in IPF patients [19]. Growing evidence illustrate that KRT8+ cells are progenitor cells derived from AT2 and further studies are needed to demonstrate their fate in animal models of lung fibrosis and their particular role in regeneration.
We identified AT2 cells with Pro-SPC marker which co-expressed KRT8, however the presence of exclusively KRT8+ cells suggests a later stage in the transition from AT2 cells to AT1 cells.These findings could confirm the literature which described the KRT8+ cells as a transitional stem cell states potentially originating from AT2 cells [12, 13, 19, 21, 22, 23].
To identify mesenchymal-like cells or intermediates of the epithelial-mesenchymal transition (EMT) process, we performed co-staining of Vimentin and KRT8 and we found cells with co-expression of both markers, suggesting that aberrant KRT8+ cells are furthermore involved in EMT in BLM rats, a phenomenon that has been described in IPF patients [30].
Recently, several studies highlighted the importance of aberrant basaloid cells in the pathogenesis and progression of IPF. To increase the understanding of their crucial role and based on the discovery of KRT8+ alveolar differentiation intermediate (ADI) cells in the mouse BLM model, we studied and demonstrated for the first time the presence of KRT8+ cells in a rat model of lung fibrosis induced by a double BLM administration. Compared to the single dose of BLM, the double administration allowed us to have a wider therapeutic window for drug testing due to the persistent fibrosis.
Besides these significant findings, there are some limitations including the big challenge to have predictive animal model IPF-like. It is known that the limited understanding of IPF pathogenesis and the heterogeneity of the disease drive the absence of effective pharmacological therapies and that the attrition rate in clinical trials is very high. For these reasons it is necessary to improve the translational predictivity and the robustness of preclinical models. None of the BLM rodent models can completely recapitulate all the IPF features as the usual interstitial pneumonia (UIP) pattern, the presence of fibroblastic foci or the typical “honeycomb”. Moreover, it has been demonstrated that they do not progresses over time, undergoing spontaneously to a self-resolution of fibrosis which nevertheless offer the opportunity to study the natural resolution of fibrosis. Despite all the challenges, the use of the second rodent species could represent an advantage in obtaining robust data of efficacious therapies in the drug discovery process to better predict successful clinical trials.
In conclusion, our results make an important contribution to the field of preclinical research, particularly by providing the possibility to test new potential compounds in two animal species, resembling the alveolar epithelial dysfunction, which both are characterized by the presence of the interesting KRT8+ cell population whose role remain to elucidate. Further studies are needed to deepen our understanding of the role of KRT8+ cells and their involvement in the key mechanisms of fibrosis and regeneration as well the aberrant persistence of regenerative intermediate stem cell states in human IPF. A second 4-week time course experiment in BLM-treated rats will include earlier time points with the aim to study the expression of the basal marker KRT8 over time and to understand if the presence of aberrant basaloid cells may represent the ineffectual epithelial regeneration which is believed to promote fibrogenesis. Understanding the specific role of subcluster of epithelial cell population, testing new effective treatment that target KRT8+ cells and modulate their deregulated pathways would represent a new approach for IPF therapies.
IPF, idiopathic pulmonary fibrosis; BLM, bleomycin; SAL, saline;
The data produced during the current study are available from the corresponding author on reasonable request.
Study design: EB, FR, VP, PC, SB, MT. Performance of the experiments: EB, FR, VP, PC, SB, MC. Data acquisition and analysis: EB, FR, SP. Manuscript drafting: EB, FR, VP, MT. All authors read and approved the final manuscript. All authors have participated sufficiently in the work to take public responsibility for appropriate portions of the content and agreed to be accountable for all aspects of the work in ensuring that questions related to its accuracy or integrity. All authors contributed to editorial changes in the manuscript.
All the procedures involving animals were reviewed, approved and authorized by the Italian Public Health System for Animal Health and Food Safety within the Italian Ministry of Health (MoH) (authorization number 246/2021-PR). All the procedures were performed within a certified animal facility: AAALAC (American Association for Accreditation of Laboratory Animal Care, https://www.aaalac.org/). Experiments were performed in full compliance with the European ethics standards in conformity with directive 2010/63/EU, Italian D. Lgs 26/2014, the revised ‘Guide for the Care and Use of Laboratory Animals’ (Guide for the Care and Use of Laboratory Animals, 1996), and the ARRIVE guidelines (Animal Research: Reporting of In Vivo Experiments).
Not applicable.
This work was funded by Chiesi Farmaceutici S.p.A.
Francesca Ruscitti, Vanessa Pitozzi, Silvia Pontis, Paola L. Caruso, Sofia Beghi, Marcello Trevisani are employees of Chiesi Farmaceutici S.p.A, the judgments in data interpretation and writing were not influenced by this relationship. All the remaining authors have not actual and perceived conflicts of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.