



 , Simona Giurdanella 4,†, Paola Rapisarda 5,†, Giulia Leotta 4,†, Antonio Cristaldi 1,2, Claudia Favara 5, Margherita Ferrante 1,2,3
, Simona Giurdanella 4,†, Paola Rapisarda 5,†, Giulia Leotta 4,†, Antonio Cristaldi 1,2, Claudia Favara 5, Margherita Ferrante 1,2,31 Department of Laboratory of Environmental and Food Hygiene, Department of Medical, Surgical Sciences and Advanced Technologies “GF Ingrassia”, University of Catania, 95123 Catania, Italy
2 International Society of Doctors for Environment ISDE Italia, Section of Catania, 95100 Catania, Italy
3 Research Center in Nanomedicine and Pharmaceutical Nanotechnology, University of Catania, 95123 Catania, Italy
4 School of Doctor specialization in Preventive Medicine and Public Health, University of Catania, 95123 Catania, Italy
5 Department of Biological, Geological and Environmental Sciences, University of Catania, 95100 Catania, Italy
†These authors contributed equally.
Abstract
Background: The six Platinum group metal elements (PGEs) comprising Ruthenium, Rhodium, Palladium, Platinum, Iridium and Osmium are grouped together in the periodic table. Human activities are mostly responsible for releasing PGEs into the environment. This systematic review focused on three PGEs with the greatest anthropogenic use, including in vehicle catalytic converters: Platinum (Pt), Palladium (Pd), and Rhodium (Rh). Consequently, these represent the greatest contributors to environmental pollution. The current review of in vivo toxicological studies (mammalian models) and in vitro cell exposure studies examined the potential harmful effects of these metalloids to mammalians, and their possible toxicity to human health. Methods: We applied Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) methodology to conduct a comprehensive search and evaluation of records in the available literature published between 01/01/2009 and 01/15/2024 in four databases. PROSPERO code ID: CRD42024471558. Results concerning the health effects of PGEs were extracted from articles according to the inclusion and exclusion criteria. After screening the records for eligibility, 22 studies were included in the final analysis. Results: This systematic review revealed that airborne PGEs significantly increased the activation of pathologic pathways in several human organs and/or perturbed various metabolic pathways. In view of the known pro-inflammatory and organ-degenerative effects of PGEs, the paucity of studies on the effect of PGEs on the central nervous system and on possible correlations with neurodegenerative diseases were particularly evident. Conclusions: The clinical complexity and chronic nature of PGE-related pathologies indicate that targeted research is essential. In light of the increasing incidence of non-communicable diseases, particular attention should be paid to the design of epidemiological studies and to environmental monitoring services.
Graphical Abstract
Keywords
- PGE toxicology
- in vivo model
- in vitro model
- mammalian
- air pollution
- PRISMA
Platinum group metal elements (PGEs) comprise Ruthenium, Rhodium, Palladium, Platinum, Iridium, and Osmium. These are primarily introduced into the environment through human activities such as industrial processes, road traffic, agricultural practices, as well as natural phenomena such as volcanic emissions [1].
This study will focus on Platinum (Pt), Palladium (Pd), and Rhodium (Rh). Due to their extensive anthropogenic utilization, these PGEs are significant contributors to environmental pollution.
Pt, Pd, and Rh have found widespread application in various industrial sectors. Pt and Pd are utilized in catalytic converters for automobiles, petroleum refining, pesticide production, cleaning agents, paints, polymers, electronics, medical devices, and pharmaceuticals [2]. Rh is crucial in the manufacture of nitric acid, glass (including liquid crystal displays), optical instruments, and nuclear reactor components [3].
Automotive catalytic converters constitute the primary anthropogenic source of PGEs. The catalytic converter, also known as the catalytic muffler, is incorporated into the exhaust system of internal combustion engines and facilitates the catalytic conversion of pollutants such as carbon monoxide (CO), nitrogen oxides (NOx), and hydrocarbons into less harmful substances [4]. Pollutants generated by combustion motors enter into contact with Pt, Pd, and Rh elements on catalytic surfaces, leading to their complete oxidation and reduction and hence the emission of less toxic exhaust pollutants. During the driving of a vehicle, PGE particles on the catalytic surface undergo both thermal mobilization and mechanical abrasion in proportion to the catalyst’s wear, before being released into the atmosphere [5].
Although the use of catalytic converters has improved air quality, they are now the primary source of Pt, Pd, and Rh emissions in the environment, including PM2.5 (atmospheric particulate matter) [6], due to the abrasion of catalytic converter surfaces [7]. Mammals, including humans, are exposed to PGEs through inhalation, dermal contact, and ingestion. The absorption and bioaccumulation of these metals in plants and animals, along with their potential toxicity, depend on various factors such as their concentration, particle size and water solubility, as well as the exposure route and environmental conditions [8]. PGEs represent a potential threat to human health [9], as evidenced by their cytotoxic, mutagenic, and carcinogenic effects [10, 11, 12, 13, 14]. In particular, nanoparticulate PGEs are a major concern due to their environmental and human impacts. PGE nanoparticles have high toxicological potential [15] due to their elevated chemical and thermal persistence. They participate in various biochemical processes as catalysts and increase absorption and oxidative interference in various human tissues and systems. There is ongoing debate regarding the toxicity of emitted PGEs to living organisms and humans, especially with regard to individuals living in urban areas or along major highways [16]. The goal of this review is to comprehensively assess all in vivotoxicological studies of PGEs using mammalian models, as well as in vitro cell exposure experiments relating to the potential health risks of PGEs. By evaluating the harmful effects of these metalloids, used primarily in vehicle catalytic converters, this review aims to shed light on their implications for human health.
The Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) approach was applied to systematically search and evaluate the literature record [17]. Searches were performed in the PubMed, Web of Science, Cochrane, and Scopus databases on articles published between 01/01/2009 and 01/15/2024. Hence, the most recent scientific research on the subject was reviewed in order to evaluate the most up-to-date information available on the toxicological implications of PGEs in mammalian models. Relevant articles in English and with full-text availability were identified in the selected databases using the following keywords and combinations thereof: “Platinum group elements and health — Platinum group toxicity — Platinum and health — Palladium and health — Rhodium and health — Palladium and platinum and rhodium and health — Catalytic converter metals and health — Pd or Pt or Rh burden disease”. Data concerning the PGEs and their impact on human health were extracted from these articles based on predefined inclusion and exclusion criteria.
The inclusion criteria were as follows: (1) studies that included a general mammalian population of both sexes, including humans; (2) studies that investigated the health effects resulting from exposure to PGEs and which also assessed their presence in atmospheric particulate matter (PM10 and PM2.5) or air samples; (3) in vitro or in vivo studies conducted using mammalian cells or live animals exposed to PEGs in urban air samples; (4) cohort studies and case-control studies.
The exclusion criteria were as follows: (1) studies that included exposed workers; (2) studies that included populations already suffering from chronic diseases; (3) studies where the full text of articles was not available; (4) review articles, surveys, meta-analyses, case reports, case series, comments, letters, and conference abstracts or posters, PhD theses.
This systematic review project was recorded in PROSPERO (International Prospective Register of Systematic Reviews) with the code ID: CRD42024471558.
The PRISMA workflow diagram outlining the screening and selection process is presented in Fig. 1. A total of 28,881 bibliographic studies were initially identified, which reduced to 4413 after removing duplicates. Subsequent screening of the remaining studies involved assessing titles and abstracts, resulting in 7644 selections, and totaling 16,824 after compilation. After excluding irrelevant studies, the remaining articles were comprehensively evaluated through full-text reading. Some studies were subsequently excluded, leading to the inclusion of 22 studies in the final analysis. These comprised 10 in vivo studies [18, 19, 20, 21, 22, 23, 24, 25, 26, 27] and 12 in vitro studies [28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39] (Table 1, Ref. [18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39]).
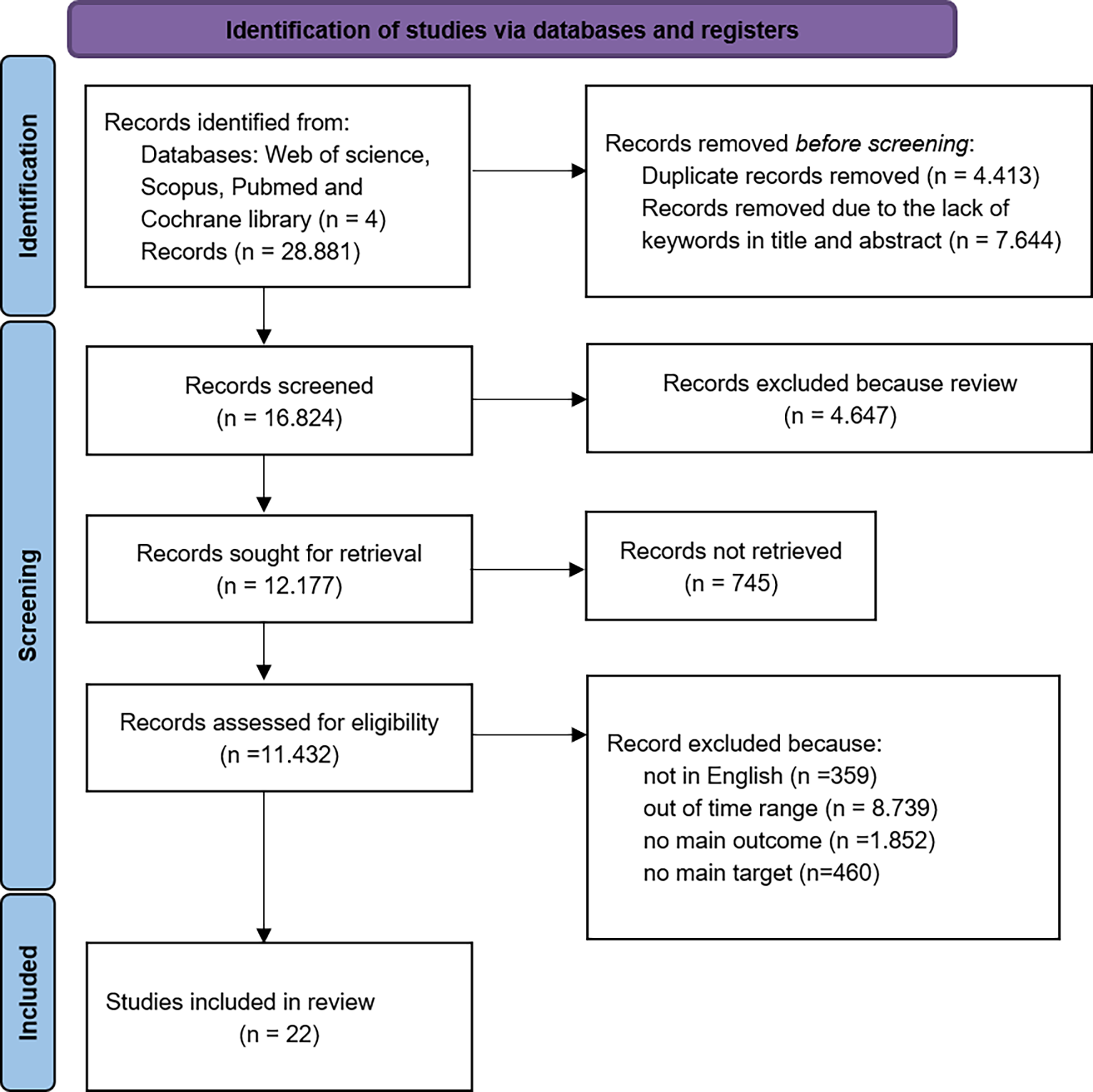 Fig. 1.
Fig. 1.
PRISMA flowchart. PRISMA, Preferred Reporting Items for Systematic Reviews and Meta-Analyses.
| Author | Element | Sample | Exposure parameters and modality of administration | Aim of study | Statistical method | LoD/LoQ | Results |
| [18] | Rhodium chloride hydrate | 35 female Wistar rats | Oral in mg/L for 14 days (0; 0.001; 0.01; 0.1; 0.25; 0.5; 1) | Effect on the immune system | SPSS | N.A. | Rh→↓IL-1b (at 0.001 mg/L) |
| ANOVA | Rh→↓IL-4 (at 0.001, 0.01, 0.25 mg/L) | ||||||
| Dunnett t test | Rh→↓IL-6 (at range 0.001–0.25 mg/L) | ||||||
| p value |
Rh→↓GM-CSF (at range 0.001–0.25 mg/L) | ||||||
| Rh→↓IL-10 (at 0.001, 0.01 mg/L) | |||||||
| [20] | PdNPs | 20 female Wistar rats | intravenous injection µg/kg (0; 0.012; 0.12; 1.2; 12) | effects of PdNPs on the immune system | SPSS | N.A. | PdNPs → ↑IL-1 |
| 10 |
ANOVA | (at 12 µg/kg) | |||||
| Dunnett t test | |||||||
| p value |
|||||||
| [21] | PdNPs | 25 female Wistar rats | repeated intravenous injections | effects on the immune system | SPSS | N.A. | PdNPs→ ↓ IL-1a, IL-4, IL-10, IL-12, GM-CSF |
| 10 |
(day 1, 30, and 60) | Dunnett t test | |||||
| mg/Kg (0; 0.012; 0.12; 1.2; 12) | p-value | ||||||
| [28] | palladium chloride PdCl2 | 30 hearts of Wistar albino male rats | perfused for 30, 60, 90, 120 minute. | toxicity on the isolated heart | Method of least squares | N.A. | PdCl2→↓ DLVP |
| trans-dichlorobis[triethanolamine-N] Pd complex (transPdCl2[TEA]2) | PdCl2 nM/L: | ↓ MBP | |||||
| (56; 560; 5600; 56,000; | ↓ HR | ||||||
| 560,000) | |||||||
| transPdCl2[TEA]2 nM/L | |||||||
| (21; 210; 2100; 21,000; | |||||||
| 210,000). | |||||||
| [22] | PdNPs | 25 female Wistar rats | intravenous | renal toxicity of PdNPs | IBM SPSS | LoD | PdNPs →↑RBP |
| 10 |
µg/kg: (0; 0.012; 0.12; 1.2; 12) | Kolmogorov–Smirnov Z-test | 0.008 mg/L | ↑b2microglobulina | |||
| Levene test | (at 12 mg/kg) | ||||||
| ANOVA | |||||||
| Dunnett’s t test | |||||||
| p value |
|||||||
| [19] | Rh chloride hydrate | 35 female Wistar rats | Oral for 14th day | toxicological effects of Rh on the kidneys | Software SPSS | N.A. | Rh chloride hydrate |
| mg L-1: (0; 0.001; 0.01; 0.1; | Kolmogorov–Smirnov Z test | from 0.1 to 1 mg L-1 → ↑RBP | |||||
| 0.25; 0.5) | ANOVA | 1 mg L-1→ ↑b2microglobulina | |||||
| Dunnett t test | |||||||
| p value |
|||||||
| [23] | PdNPs | 20 female Wistar rats | Intravenous for 14 days | effects of on the reproductive system | IBM SPSS | N.A. | PdNPs 12 µg/kg → |
| 10 |
µg/kg: (0; 0.12; 1.2; 12) | Kolmogorov–Smirnov Z test | ↓E2 | ||||
| Levene test | ↑LH | ||||||
| Dunnett t test | ↓T | ||||||
| p value |
|||||||
| [24] | PdNPs | 20 female Wistar rats | Intravenous | impact on the HPG-axis | SPSS | N.A. | PdNPs 0.12, 1.2, 12 → ↑FSH |
| 10 |
at days 1, 30, and 60: | ANOVA | |||||
| µg/kg: (0; 0.12; 1.2; 12) | Levine’s test | ||||||
| Dunnett t test | |||||||
| p value |
|||||||
| [27] | PtNPs | 12 Wistar rats | Oral for 30 days, | toxicity and cellular injury in organs: heart, kidney, liver | ANOVA | N.A. | PtNP → |
| 12 |
mg/kg; (0; 10; 50; 100) | Duncan multiple range test | ↓liver, kidney, heart weight loss | ||||
| p value |
↓ albumin | ||||||
| ↑atherogenic index | |||||||
| ↑heart, liver, kidney proteins | |||||||
| ↑creatinine | |||||||
| ↑bilirubin serum | |||||||
| ↑ALT in liver | |||||||
| In all the organs PtNPs 10 mg/kg: | |||||||
| ↑ inflammation and intercellular cracking. | |||||||
| PtNPs 50, 100 mg/kg ↑inflammation and cellular degeneration. | |||||||
| [26] | Pt, Pd, Rh | 25 female rats | Oral for 8 week | hematological effects | ANOVA | N.A. | PGM →↑Monocytes ↑significant immunostimulation at 0.1 ppm Pt. |
| (Water-soluble Pt | ppm: (0; 0.1; 1.0; 5.0; 10.0) | Student–Newman–Keuls test | |||||
| in the form of hexachloroplatinic acid (H2 [PtCl6].6H2O), | PGM mix solution: 0.1 or 1.0 ppm | p value | |||||
| Pd (II) in the form of PdCl2, | |||||||
| Rh(III) in the form of RhCl3) | |||||||
| In vitro | |||||||
| [30] | PdNPs (10 |
Rat-1 cells | Cells cultures with PdNPs concentrations: | toxicity and cellular injury associated with PdNPs acute and subacute exposure. | ANOVA | N.A. | PdNPs (1–2 µg/mL for 120 h) → |
| A549 cells | (0; 1; 2 µg/mL) | Bonferroni post hoc comparisons tests. | ↓ growth of Rat-1 | ||||
| cultures analyzed at 0, 24, 72, 120 h after preparation | p value |
PdNPs (2 µg/mL from 96 to 120 h) | |||||
| ↓ growth of A549 cells. | |||||||
| [33] | PdNPs | - PBEC cells | Cell cultures Pd NP-treated in 24 h with concentrations of: (0.01; 0.1; 1; 10 µg/mL) to evaluate PGE2 and IL8 production; (10; 25 µg/mL) to evaluate apoptosis. | PdNPs interferences on cells viability, relatively to: | Wilcoxon signed rank test. | N.A. | PdNPs (10 µg/mL after 2 h) → absorption in PBEC cells within endosomes. |
| (10.4 |
- A549 cells | Addition of TNF- |
- absorption | p value |
PdNPs (up to 10 µg/mL) → | ||
| -vital dye exclusion, | nonlinear trend IL-8 ↓ PGE2 both cellular secretion | ||||||
| -apoptosis. | -↓ reactivity of PBEC to TNF- | ||||||
| -release of soluble biomarkers IL-8 and PGE2 | PdNPs up to 10 µg/mL (in PBEC markedly) → | ||||||
| ↓ cell viability. ↑induction of apoptosis (DNA fragmentation) ↑caspase activation | |||||||
| [38] | PdNPs (5–10 nm) | PBMC cells from 20 healthy female volunteers’ blood samples: | PBMC cultures incubated overnight at 37 °C with PdNPs or PdIV salt (0; 10-5 M) with and without LPS (10 µg/mL) | Immune potential of PdNPs in PBMC cytokines releasing compared to PdIV salt. | SPSS | N.A. | In nonatopic samples: |
| PdIV salt (hexachloropalladate salts) | -12 nonatopic | Kolmogorov-Smirnov test | PdIV → ↓ IL-10, IL-17 without LPS; ↓IFN- | ||||
| - 8 Pd atopic | Wilcoxon ranked sum test | In Pd atopic samples: | |||||
| PdIV → ↓ IL-10 with and without LPS; PdNPs → ↓ IL-10 without LPS, ↓ TNF- | |||||||
| [39] | -PdNPs (5–10 nm) | PBMC cells from 8 healthy female nonatopic volunteers’ blood samples | PBMC cultures incubated with PdNPs or PdIV salt (0; 10-5 M; 10-6 M) with and without LPS (10 µg/mL) | PdNPs immune potential in PBMC cytokines releasing (IL-5, IL-10, TNF- |
SPSS | N.A. | In cell cultures without LPS: |
| - PdIV salt | Kolmogorov-Smirnov test | - PdIV salt (dose-related way) → ↓IL-10, IL-17 | |||||
| (hexachloropalladate salts) | Wilcoxon test | In cell cultures with LPS: | |||||
| - PdIV salt → ↓IL-10, IL-17, IFN- | |||||||
| Pd NPs→ ↓TNF- | |||||||
| [35] | PtNPs (5.8 nm; 57 nm) coated with polyvinylpyrrolidone | NHEKs from 3 adult donors | NHEK incubated for 24 and 48 hours with two sizes PtNPs (6.25; 12.5; 25 µg/mL) | PtNPs cytotoxicity, genotoxicity, morphological, metabolic changes, activation of cellular signaling pathways. | Student’s t-test | N.A. | PtNPs both sizes→ ↓ cellular metabolism; no effects on cell viability/migration. |
| Tukey’s rank-invariant resampling test. | 5.8 nm PtNPs→ ↓ DNA stability; alterations in apoptosis activity of caspase 9 and caspase 3/7 | ||||||
| p value |
|||||||
| [32] | PtNPs ( |
Spermatozoa from 5 hetero-zoospermic ejaculate samples of adult New Zealand white rabbits. | Spermatozoa incubated in 0.9% NaCl with different concentrations of PtNPs (0; 62.5; 31.25; 15.63; 7.81; 3.91; 1.95; 0.98; 0.49; 0.24; 0.98; 0.49; 0.24 µg/mL) at time intervals: 0, 2, 4, 6, 9, 24 hours. | effects on male reproductive system by analyzing spermatozoa parameters: MOT, %, PRO, %, VCL µm/s, viability. | GraphPad Prism 8 | N.A. | PtNPs → ↓spermatozoa MOT, %, PRO, %, VCL µm/s (time and dose-dependent effect) |
| ANOVA | ↑slight increase in MOT and PRO at initial time interval. | ||||||
| Dunnett’s test | No viability affection detected. | ||||||
| *p value |
|||||||
| [36] | PdNPs Pd (II) ions | PBLs from three healthy donors | cells exposed to different concentration of Pd-NPs (0, 0.01, 0.1, 1, 10, 50, 100, 200 µg/mL) or Pd ions (0, 0.01, 0.1, 1, 10, 50, 100, 200 µg/mL). | effects of PdNPs and Pd (II) ions on cell apoptosis, oxidative stress and cell cycle arrest. | ANOVA | N.A. | PdNPs and Pd (II) ions → ↓ PBLs growth (dose-dependent way); higher inhibition by Pd (II) ions. |
| p value |
PdNPs and Pd (II) ions →↑cytotoxicity and apoptosis through: ROS enhancement (markedly in Pd (II) ions) and cell cycle arrest in G1 stage. | ||||||
| [25] | snPt1 ( |
BALB/c and C57BL/6 male mice | snPt1 or snPt8 Injection (twice weekly for 4 weeks). | effects of snPt1 and snPt8 on tissues (kidney, spleen, lung, heart, and liver) after single- and multi-dose administration. | Student’s t test. | N.A. | snPt1 single/multiple intravenous/intraperitoneal (10 mg/kg) administration →↑nephrotoxicity, impaired renal function (BUN levels↑), tubular atrophy, kidney inflammatory cell accumulation |
| snPt8 ( |
Number N.A. | BALB/c: intravenously (5 to 20 mg/kg body weight) | p value |
snPt1 single intravenous administration (10 mg/kg) → ↑liver vacuole degeneration | |||
| C57BL/6: intraperitoneally (10 mg/kg body weight) | snPt8 single/multiple intravenous/intraperitoneal administration → No renal cytotoxicity. or hepatotoxicity | ||||||
| Control: C57BL/6 intraperitoneally with equivalent volume of vehicle (water). | |||||||
| [31] | PdNPs ( |
J774 murine cells | J774 cells incubated for 24 hours with PdNPs at different concentrations (25, 100, 200, 300, 400, and 500 µg mL-1) | PdNPs hazardous effect on murine macrophages, cell viability and proliferation. | Student’s t-test | N.A. | PdNPs (at 400 and 500 µg mL-1 after 6 hours) → ↓ cell viability, ↑apoptosis (time and dose-dependent way) ↑ROS generation |
| p value | |||||||
| [29] | Rh (III) salt | Rat-1 cells | Rat-1 incubated with Rh (III) salt (0.1 or 0.3 mM) in 24 h; (0.05 to 0.6 mM) in 48 h. | Rh (III) salt cyto-toxic and bioactive effects in fibroblasts. | ANOVA | N.A. | Rh (III) salt (dose and time- dependent) → ↓ cell growth, specifically: |
| Bonferroni post hoc multiple comparison tests. | IC50 at 48 h (0.3 mM); | ||||||
| SPSS | cell cycle alterations: ↓ cells in the G0/G1 phase, ↑ cells arrested in the S and G2/M phases (correlateable to ↓ DNA synthesis/chromosome condensation) | ||||||
| p value |
↑apoptotic cells | ||||||
| ↑oxidative stress (↑intracellular ROS) | |||||||
| ↑p21 ↑Waf1 and ↑p27Kip1, ↓cyclin D1, ↑ pRb protein (evident at the lowest dose: 0.05 mM). | |||||||
| [37] | PtNPs | cultured neonatal mice ventricular cardiomyocytes | In vitro for 5 minute | electrophysiological toxicity of PtNPs | Student’s t-test | N.A. | PtNPs (5 and 70) → |
| 5 nm | adult male C57BL6/J mice | g/mL: (10-9 – 10-5) | ANOVA | ↓the densities of IK1, Ina and Ito channels | |||
| 70 nm | in vivo intravenous infusion | p value |
In vivo, PtNPs → | ||||
| mg/kg (3–10) | ↓ the HR | ||||||
| and induced AVB at 10 mg/Kg | |||||||
| [34] | PdNPs (37 nm) | BEAS-2B cells | BEAS-2B exposed through ALI system 0.5, 1 or 2 h at (214 mL/min speed); assays after: 24, 48 h, 7 days after reseeding. | effects of PdNPs exposure in 3 cells models (lung, vascular, coagulation); viable cell counts and RNA analysis | ANOVA | N.A. | In BEAS-2B cell PdNPs (2 h exposure). → |
| 10 mcg/mL | HCMEC cells | HCMECs exposed to PdNPs (0, 5 µg/mL) incubated 30 min in culture chambers. | Tukey’s multiple comparison test | ↓ cell growth (detectable already after 24 h), down to 50% of the cell number at 48 h. Time dependent effect. | |||
| 1.5 mL Fresh human whole blood | Blood samples incubated under constant rotation for 60 min at 37 °C treated in 150 mM NaCl with PdNPs (0; 10 µg/mL). | *p value |
↑ apoptosis and cell cycle arrest ↑p53 ↑CDKN1A. | ||||
| TiO2 NPs (10 µg/mL) used as positive control. | In HCMEC cell PdNPs → ↓ live/dead assay ↑ P-selectin upregulation (↑thrombo-inflammatory process) | ||||||
| In blood samples PdNPs→↑ coagulation through: ↑ activation of platelets (measured as ↓ 45% platelet number) | |||||||
| ↑ TAT complexes formation after Pd blood contact | |||||||
| = FXII bound to both PdNPs and TiO2NPs, FXII activates in FXIIa when bound to TiO2) | |||||||
| ↑inflammation via activation of KKS→Bradykinin FXIIa |
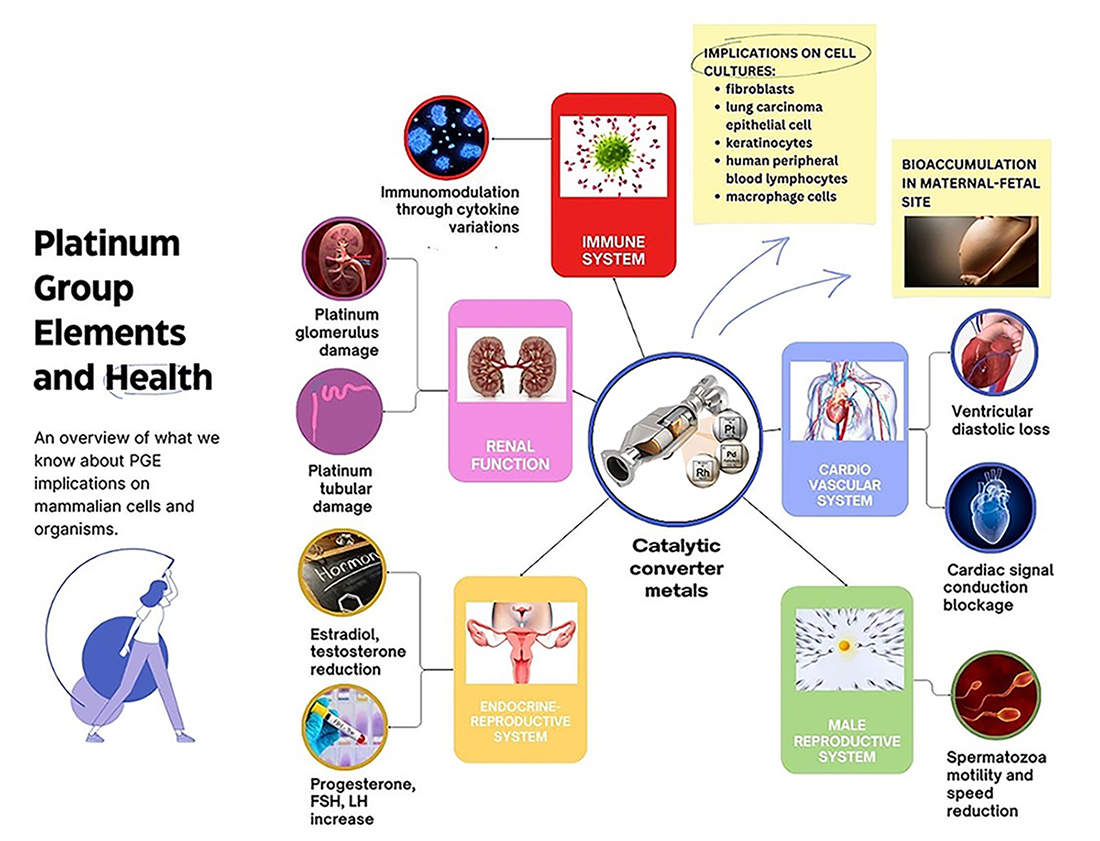
The findings relating to exposure to PGEs were categorized into two subsections: organ health effects and in vitro cell health effects.
The eligible records included in vivo studies that focused on the pharmacokinetics and pharmacodynamics of PGEs in mammalian organisms. These considered exposure to different concentrations at the systemic level [18, 19, 20, 21, 22, 23, 24, 25, 26, 27], or direct administration into specific organs via intra-organ injection [28]. Implications for specific body systems were examined, in particular for the immune, renal, endocrine-reproductive, and cardiovascular systems. The eligible records included in vitro studies that evaluated the impact of exposure to PGEs using nanoparticles of the same size as those emitted by vehicle catalytic converters. Experiments were conducted using various mammalian cell culture models, including neoplastic lung fibroblast and epithelial cells [29, 30], macrophages [31], sperm cells [32], as well as human cell cultures of lung [33], endothelial, hematological and epithelial cells [34], keratinocytes [35], lymphocytes [36], cardiomyocytes [37], and peripheral blood samples [38, 39].
The findings from 7 studies (3 in vivo [18, 20, 21], 4 in vitro [31, 36, 38, 39]) on the effects of PGEs on the immune system are presented.
To study the impact of Rh on the immune system, Iavicoli et al. [18]
exposed 35 female Wistar rats to six different doses of rhodium hydrate chloride
(0.001, 0.01, 0.1, 0.25, 0.5, and 1 µg/L) for 14 days (sub-acute
exposure). All cytokine values (IL-1
Iavicoli et al. [20, 21, 40] also investigated the potential immune
implications of Pd nanoparticles (PdNPs) in a murine model. They had previously
reported that sub-chronic exposure to Pd (90 days) influenced the serum levels of
IL-2 and INF-
Newkirk et al. [26] used a murine model to analyze the activity of immune cells after 8 weeks of daily exposure to different concentrations (0.1, 1.0, 10.0 ppm) of water-soluble Pt (IV) in the form of hexachloroplatinic acid (H2[PtCl6].6H2O). Increasing Pt concentrations altered the immune response and leukocyte quantity, as inferred from the differential white blood cell count. Monocytes showed an increasing trend at all concentrations of Pt exposure, with significant immunostimulation at 0.1 ppm of Pt. Monocytes are responsible for necroptosis, phagocytosis, necrophagocytosis and chemotaxis via the induction of cytokine production. Of the cell lines investigated, only monocytes showed a statistically significant difference compared to the controls. Various in vitro studies have investigated the immunotoxicity of PGEs by exposing various immune cell lines to PGE xenobiotics and then evaluating for potential cytotoxic alterations. Pd compounds are widely recognized in the medical literature [41] as being more immunotoxic compared to Pt compounds and Rh salts. In vitro studies by Boscolo et al. [39] and Reale et al. [38] examined the potential immunotoxicity of Pd. Both groups investigated the immunological potential of Pd in the form of PdNP (5–10 nm size) and potassium hexachloropalladate at doses of 10-5 M or 10-6 M on peripheral blood mononuclear cells (PBMCs). These were obtained from 8 healthy non-atopic women in the study by Boscolo et al. [39], and in 8 atopic women and 12 non-atopic women in the study by Reale et al. [38]. The incubation of PBMCs was carried out in the presence or absence of immunostimulant, specifically lipopolysaccharide (LPS), to evaluate any interference by the Pd xenobiotics in relation to external immune activation by LPS.
The Boscolo et al. [39] and Reale et al. [38] studies reported the observations described below.
In the non-atopic population, Pd salt inhibited the release of cytokines (IL-10
and IL-17) in cell cultures without LPS, while in immunostimulated cultures Pd
salt significantly increased IFN-
Zivari Fard et al. [36] investigated the proapoptotic effects of PdNPs on human lymphocytic cells. Human peripheral blood lymphocytes isolated from healthy donors were exposed to different concentrations of PdNPs (0, 0.01, 0.1, 1, 10, 50, 100, 200 µg/mL; 15 nm size) and Pd ions (0, 0.01, 0.1, 1, 10, 50, 100, 200 µg/mL). After 24 h incubation, PdNPs and Pd ions inhibited the growth of lymphocytes in a dose-dependent manner. In addition, they enhanced cell cytotoxicity by increasing the level of reactive oxygen species (ROS) and causing cell cycle arrest in the subG1 phase. Pd ions induced a higher level of apoptosis and ROS than PdNPs.
Aarzoo et al. [31] also reported the same cytotoxic mechanisms of apoptosis and ROS generation in the murine J774 macrophage immune cell line following exposure to bioengineered PdNPs. The cells were incubated with various concentrations (25, 100, 200, 300, 400, and 500 µg/mL; 4 nm size) of PdNPs for 24 h. Reduced cell vitality, increased apoptosis, and significant generation of free radicals was already apparent after a 6 h incubation period, especially at concentrations of 400 and 500 µg/mL.
Fig. 2 shows that PGEs can play an immunomodulatory role, implying both pro- and anti-inflammatory effects [14, 16, 17, 22].
 Fig. 2.
Fig. 2.
Platinum group metal elements (PGEs) and the Immune System: Evidence of immunostimulation/immunosuppression following the exposure of Wistar rats to PGEs. Depending on the mode of administration, PGEs such as Pd can play an immunomodulatory role, implying both pro- and anti-inflammatory effects.
The kidneys play a crucial role in eliminating metabolic byproducts from the body. They eliminate toxins and pollutants from the bloodstream, including environmental chemicals such as PGEs. However, this excretory function makes them susceptible to potentially adverse effects due to prolonged exposure to toxic substances [42].
Potential renal toxic effects due to PGE exposure have been reported in the literature, including histopathological analyses in experimental murine models [19, 22, 25, 27].
Following intravenous (5, 10, 15, and 20 mg/kg;
Both Fontana et al. [22] and Iavicoli et al. [19] studied the effects of PdNPs on renal function. In the study by Fontana et al. [22], female Wistar rats were exposed to PdNPs via intravenous administration, with the concentrations used being compatible to inhaled Pd levels in the air. In contrast, Iavicoli et al. [19] used drinking solutions to achieve controlled exposure. Higher doses (1.2 and 12 µg/kg) simulated potential occupational Pd exposure, whereas lower doses (0.012 and 0.12 µg/kg) allowed the study of potential adverse effects at exposure levels similar to those experienced by the general population. At the highest treatment dose (12 µg/kg), elevated levels of urinary albumin and retinol binding protein (RBP) were observed. Electron microscopy observations confirmed the suggestion of tubular nephrotoxicity.
Iavicoli et al. [19] also examined the effects of Rh toxicity on renal
function. The ingestion of Rh chloride hydrate solutions at varying doses by
female Wistar rats resulted in a significant increase in the urinary level of RBP
and a trend for increased
Adeyemi et al. [27] investigated the cardiotoxicological implications of Pt exposure by exposing murine models to orally administered PtNPs for 30 days. Biochemical-enzymatic examinations and histopathological analyses of organs such as the liver, kidney and heart revealed significant increases in the atherogenic index at concentrations of 10 and 50 mg/kg. Histopathological examination revealed a dose-dependent reduction in cardiac weight, degeneration of the cardiac tissue and inflammation. The cardiotoxicological implications of PGEs also involve cardiac electrophysiology. Lin et al. [37] examined the acute toxic effects of PtNP (5 and 70 nm sizes) on cardiac electrophysiology. Their results suggest that PtNP may induce a potentially hazardous block in the conduction of cardiac signals.
Few studies have examined the effects of Pd on the heart. Perić et al. [28] assessed the toxicity of organic (trans-dichlorobis[triethanolamine-N] Pd complex) and inorganic (palladium chloride) Pd compounds injected into the isolated hearts of Wistar albino rats. The results indicated that inorganic Pd compounds caused a clear reduction of cardiac contractility, leading to lower left ventricular diastolic and mean pressure and decreased heart rate. Organic Pd compounds did not show a significant impact on cardiotoxicity [28].
Studies conducted by Leso et al. in 2018 [23] and 2019 [24] examined the effects of acute (14-day) and sub-chronic (1, 30, and 60-day) exposure to PdNPs in female Wistar rats. These authors assessed changes in serum sex hormone concentrations, including estradiol (E), follicle-stimulating hormone (FSH), luteinizing hormone (LH), progesterone (P) and testosterone (T). Acute exposure to PdNPs resulted in dose-dependent increases in FSH, LH and P. Significant reductions in E and LH levels were observed even at the lowest exposure doses, while the reduction in T was significant at the highest exposure dose. Sub-chronic exposure for 90 days resulted in significantly higher average levels of FSH in PdNP-treated groups compared to controls [23, 24].
Results from the literature indicate that human seminal glands are particularly vulnerable to the presence of xenobiotics, and in particular to trace metal elements. These effects can occur even at low levels, with the metals acting as cofactors in crucial enzymatic reactions for spermatogenesis, spermatozoa motility, and capacitation [43]. However, the influence of PGE metals on the genesis and maturation of sperm is still poorly understood, and there is no data regarding the toxicity of PGEs on sperm in the human seminal model. Dianová et al. [32] investigated the effects of PtNPs (0.98 µg/mL, 1.95 µg/mL, 3.91 µg/mL, 7.81 µg/mL, 15.63 µg/mL, 31.25 µg/mL) on rabbit seminal samples. PtNPs were found to negatively influence sperm parameters such as motility and speed, especially at higher concentrations and after chronic exposure. Sperm vitality was the only parameter that remained relatively stable.
Limited data are available regarding the in vitro cellular effects of PGEs and their toxicity.
Iavicoli et al. [30] conducted an in vitro study to assess the potential toxic effects and possible mechanism of action of PdNPs in normal rat diploid fibroblasts (Rat-1) and in the human lung carcinoma epithelial cell line A549. The latter was used to simulate the lower respiratory tract. The effects of PdNPs on cell growth, cell cycle progression, apoptosis induction, DNA damage, production of ROS, and the expression of cell cycle regulatory proteins were examined after 48, 72, 96 and 120 h incubation with increasing concentrations of PdNPs (0–3 µg/mL). Treatment with 1 µg/mL PdNPs inhibited the growth of Rat-1 cells over time, ranging from 10% after 48 h, to 70% after 120 h. At a concentration of 2 µg/mL, the inhibition of cell growth was 30% after 48 h and 80% after 120 h. A549 cells tested with the same concentrations of PdNPs showed lower toxicity. Specifically, treatment with 1 µg/mL inhibited A549 cell growth by 10% after 48 h and 30% after 120 h, while treatment with 2 µg/mL reduced A549 cell growth by 20% after 48 h and approximately 50% after 120 h. In both cell lines, inhibition of cell growth was mainly associated with the accumulation of cells in the G0/G1 phase of the cell cycle, as well as fewer cells in the S phase. These results indicate that cells are maintained in the G0 state, or prolonged/arrested in the G1 phase, and suggest that DNA damage prevents the cells from entering the S phase.
In an earlier study on Rat-1 diploid fibroblast cells, Iavicoli et al. [29] studied the effects of Rh salts (rhodium III) on the cell cycle, apoptosis, and the expression of cell cycle regulatory proteins. The Rh salts caused a dose- and time-dependent inhibition of cell growth and viability (0.3 mM for 48 h). In contrast to the 2017 study on Pd, cell cycle arrest occurred in the S and G2/M phases of the cell cycle rather than in the G0/G1 phase. This effect is likely because of inhibition of the DNA synthesis process, or to inhibition of the S to G2/M transition due to blocking of cell division activities such as chromosome condensation or spindle formation. A significant increase in apoptotic cells was also observed, accompanied by increased intracellular ROS and DNA fragmentation. Additionally, increased expression of the cell cycle regulatory proteins retinoblastoma (pRB), p21Waf1 and p27Kip1 (CDK inhibitors) was observed, as well as reduced expression of cyclin D1, which is involved in cell cycle progression. The study also found that increasing the dose and exposure time (from 0.05 to 0.6 mM for 48 h) resulted in the same trends for all proteins except pRb. The expression of cyclin E showed no changes at any of the test doses.
Wilkinson et al. [33] conducted an in vitro study to assess
apoptosis following the exposure of primary human bronchial epithelial cells
(PBEC: cells from the upper respiratory tract) and human alveolar carcinoma cells
(A549) for 24 h to PdNPs (10.4
Significant changes in the activity of caspase 9 and caspase 3/7 were also observed following the exposure of human epidermal keratinocytes to Pt for 24 h and 48 h (5.8 nm and 57 nm PtNP size; concentrations of 6.25, 12.5, and 25 µg/mL) [35]. Evidence of dose-dependence was found for cellular absorption via intracellular organelles similar to endosomes/lysosomes, genotoxic damage to DNA (starting from 12.5 µg/mL), caspase 9 activation at 24 h, and inhibition of caspase 3/7. Rapid increases (4 h) in the levels of MAPK proteins (JNK and ERK1/2 involved in cell growth regulation, metabolism, survival, and proliferation) and Akt proteins (kinases activated under stress conditions) were also observed, resulting in significantly reduced cellular metabolism. No significant differences were found in the cell cycle between quiescent (G0/G1) and proliferative (S and G2/M) phases.
In a recent study, Fromell et al. [34] evaluated the inhalation toxicity of Pd particles (37 nm average size) using three in vitro models: an Air Liquid Interface (ALI) system where BEAS 2B bronchial epithelial cells come into contact with air-borne PdNPs; an endothelial cell model; and a model of human blood used to investigate vascular and coagulative responses to PdNPs exposure. Counting of BEAS 2B bronchial epithelial cell colonies following 24 h exposure to PdNPs in the ALI model revealed cell cycle arrest (also found in [30]), early apoptosis, and up to 50% lower cell count compared to the control group. These events first appear after 0.5 h of exposure and peak 2 h after the start of exposure. The decreased cell vitality appears to be maintained, with the observations repeated 16 days later in the same cells. In the remaining two cell models (endothelial cells and whole human blood), exposure to PdNPs resulted in increased coagulative activity, specifically in terms of thrombin generation, platelet consumption, and activation of the kallikrein-kinin system. Compared to control cells treated with Titanium Dioxide nanoparticles, a potent coagulation activator, the prothrombotic and inflammatory effects of PdNPs were less aggressive, but still indicative of potential vascular damage [34].
As “primary” air pollutants and toxic “secondary” air pollutant species, PGEs may have negative health effects through transformation and uptake by organisms, or through bonds with salts or particulate matter byproducts. In this regard, the formation of halogenated PGEs complexes, which have a greater potential to induce cellular damage, can occur in the presence of chloride in lung fluid [44]. The findings of this systematic review highlight the ability of airborne PGEs to increase the activation of pathologic pathways in several human organs, and/or to perturb metabolic pathways.
The immune system is involved in maintaining tissue balance and overall system integrity through deep connections to other body systems such as metabolism, the central nervous system, and the cardiovascular system [45]. The potential negative effects of PGEs on the immune system are obviously crucial to the overall health of the organism. Anthropogenic emissions of PGE can modulate the immune response and promote allergic reactions [46]. The in vivo and in vitro studies discussed in this review have reported immunomodulatory effects of Pd. They also evaluated the potential immunotoxicity of Pt and Rh, particularly for sub-chronic exposures that represent the typical environmental conditions experienced by the general population.
None of the reviewed studies identified the exact etiopathogenesis of the immune alterations observed after Rh/Pd/Pt exposure [47, 48]. An in vivo study suggested a direct, dose-dependent toxic action of PGEs on T helper lymphocytes, as well as imbalances in Th1/Th2 immune responses [39]. Nevertheless, further studies are needed to determine the etiology of such immune alterations. The toxicity of PGEs may also affect other immune cells, including monocytes and macrophages [26]. Moreover, immunomodulation may vary according to the type of exposure, such as high-intensity exposure over a short period (acute exposure), or low-intensity exposure over a prolonged period (sub-chronic exposure). Immunostimulation can occur in response to acute exposure, and immunosuppression may occur as an adaptive response to chronic exposure. Depending on the mode of administration, PGEs like Pd can have a significant immunomodulatory role, involving both pro- and anti-inflammatory effects. The findings of this review also highlight the impacts of PGEs (inorganic PdNPs in the form of organic palladium chloride, and PtNPs) on the cardiovascular system. Cardiac impairments include inflammatory and degenerative damage to cardiac tissue [27], impaired myocardial function (loss of cardiac contractility and ventricular dysfunction during diastole) [28], and impaired cardiac electrophysiology due to the blocking of signal conduction. According to Lin et al. [37], this electrophysiological toxicity is caused by nanoscale interference with extracellular ion channels, rather than oxidative damage or other slower biological processes.
Exposure to PGEs was also reported to cause endocrine toxicity due to alterations in hormonal and endocrine mechanisms. This contributes to pathogenesis in various reproductive systems, such as early puberty, reduced fertility, tumors, and maternal-fetal outcomes in pathological gestational models. Nanoparticle exposure can affect fetal and neonatal development during the fertile age or during pregnancy, or the nanoparticles may reach the newborn via secretion into breast milk. Pregnant women may be exposed to PGEs through various routes such as drinking water, diet, indoor and outdoor pollution, and the workplace. The adsorbed pollutants can cross the placental barrier, thereby compromising its protective function and leading to the accumulation of pollutants in the fetus and in trophic and excretory organs such as amniotic fluid and placenta. This was demonstrated by Melber and colleagues [43] who intravenously injected palladium chloride in rats. The first three months of organogenesis are the most sensitive period for fetal development. During this time, exposure to trace metals can be harmful and may affect the division and differentiation of fetal cells [43]. Caserta et al. [49] reported that certain heavy metals, including Pt, can be detected in amniotic fluid during the second trimester of pregnancy (15–18 weeks), whereas Pd was not detected. Although heavy metal particles can clearly pass through the placenta, further research is needed to fully understand the fetal health outcomes associated with this exposure [50]. Stojsavljević et al. [51] analyzed the levels of metallic trace elements, including Pd, Pt and Rh, in placental tissues from 105 healthy pregnant women. The participants met specific inclusion criteria, such as age between 20 and 40 years, gestational age of 37–42 weeks, uncomplicated vaginal delivery, normal fetal weight, and an Apgar score of 9. Women with professional or incidental exposure to heavy metals and toxic substances were excluded, and the large majority of participants (90%) resided in metropolitan areas. The women in the study were healthy and had no confounding factors, thus providing a normal reference range for 50 different metals in placental tissues from a physiological scenario. These results provide important information for metal biomonitoring studies (including Rh, Pd, and Pt) in solid tissues. Exposure to PGEs also compromises fertility in the male reproductive system [32, 51]. Sperm parameters including spermatogenesis, spermatozoa motility and capacitation show weak but significant negative impacts following exposure to xenobiotics. Karabulut et al. [52] found no significant correlations with PGE metals when comparing normospermic and pathological human seminal samples. However, the study by Sharma et al. [53] found significant reductions in sperm motility in response to Pd and Pt treatment in a murine model.
Following review of the selected studies involving in vitro cell experiments and in vivo histo-anatomical pathological models, the pathogenic mechanisms of PGEs can be summarized as follows. These include apoptosis (as detected by endosomal accumulation, oxidative stress, DNA fragmentation, activation of cell cycle regulatory proteins such as caspases, cyclin D1, p21Waf1, p27Kip1), cell cycle arrest during progression towards mitosis, reduction in cellular motility and vitality, and alterations in cellular electrophysiology.
The temporal kinetics also predict mechanisms of tolerance during chronic exposure, with a reduction/stabilization in the responsiveness to xenobiotics. Maximum responsiveness was observed with acute exposure after a short time interval (0.5 to 2 h), even at the lowest concentrations tested.
Notable limitations were identified in the reviewed studies conducted on in vivo models, mostly due to ethical issues and to potential anomalies arising from interspecies differences. To address these limitations, further in vitro and in vivo investigations are required to obtain a comprehensive understanding of the effects of PGEs on various human systems, e.g., the immune system, major excretory organs like the liver, lung, kidney, the reproductive system, and the cardiovascular system.
Despite the frequently mentioned pro-inflammatory and organ-degenerative effects of PGEs, there was a notable lack of studies on the effects of PGEs on the central nervous system and on possible correlations with neurodegenerative diseases. The clinical complexity and chronic nature of these pathologies means that targeted research is essential. In view of the increasing incidence of non-communicable diseases, particular attention should be paid to the design of epidemiological studies and to environmental monitoring.
With regard to the neurotoxicity of PGEs, extensive evidence is only available for the specific application of Pt as a chemotherapeutic agent. The use of Pt in this context results in cytotoxic effects on various organs and systems, including the nervous system. These can include neurotoxicity, peripheral neuropathy, and altered neurocognition. Neurotoxicity may manifest with symptoms such as sensory neuropathy, motor neuropathy, disturbances of balance and coordination, and central nervous system dysfunction. Peripheral neuropathy is characterized by pain, tingling, numbness, and weakness in the extremities, while altered neurocognition can lead to problems with memory, concentration, and overall cognitive function. These effects may vary in severity depending on the type of Pt chemotherapy, dosage, duration of treatment, and individual patient susceptibility [54, 55].
The aim of this review was to investigate a type of environmental exposure to PGEs that is not comparable in either concentration or exposure modality to iatrogenic PGE exposure. Nonetheless, the well-known neurotoxicological implications of Pt in pharmacology may serve as a stimulus and basis for future environmental scientific research on Pt and related PGEs.
This systematic review has highlighted the toxicological impact of PGEs across multiple cellular models (fibroblasts, polymorphonuclear cells, keratinocytes, spermatozoa, lung epithelial cells) and in various multi-organ analyses (kidney, heart, liver, endocrine-reproductive system). Depending on the duration of exposure and the dose employed, the effects of PGEs can include the alteration of inflammatory and immune mechanisms via the production and release of biomarkers, induction of moderate prothrombotic effects at vascular sites, and causing nephropathic effects at tubular sites. In light of the limitations mentioned earlier, future studies should focus on evaluating long-term and low-dose effects, similar to those experienced by the general population. In particular, the effect of PGEs on reproductive health should be carefully evaluated, especially in women who are exposed during fertility or pregnancy. Moreover, it is important to gain a better understanding of bioaccumulation and maternal-fetal health in relation to metals and ubiquitous PGEs. This should help to inform more adequate healthcare planning and the implementation of preventive measures for public health. Further studies should also be performed on male fertility and PGEs. Despite the lack of clear evidence linking PGEs with sperm toxicity in human seminal models, studies with animal models suggest that PGE metals may have negative effects on sperm motility, indicating a need for further studies. Overall, the findings of this review highlight the need to monitor environmental levels of PGEs and to continue research on their bioavailability, behavior, speciation, and associated toxicity. This should enable a better assessment of their potential adverse effects on human health.
Data is available as supplementary material. All data points generated or analyzed during this study are included in this article and there are no further underlying data necessary to reproduce the results.
GOC: Conceptualization, Investigation, Writing, Revision, Supervision, Validation; SG: Writing— Reviewing and Editing, Data curation; PR: Writing, Methodology, Data curation; GL: Writing, Formal analysis; AC: Formal analysis, Visualization, Software; CF: Formal analysis, Visualization, Software; MF: Investigation, Supervision, Validation and Editing. All authors have participated sufficiently in the work to take public responsibility for appropriate portions of the content and agreed to be accountable for all aspects of the work in ensuring that questions related to its accuracy or integrity. All authors read and approved the final manuscript. All authors contributed to editorial changes in the manuscript.
Not applicable.
Not applicable.
The authors of this study benefited from funding by the AIRBOrnE project n.22358 of 19 January 2023 - Line of Intervention 3 “Starting Grant” of the “PIACERI” - University of Catania research incentive plan “2020/2022”.
The authors declare that they have no conflict of interest. Gea Oliveri Conti is serving as one of the Editorial Board members of this journal. We declare that Gea Oliveri Conti had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Antoni Camins.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.