
 , Runzhi Chen 3,*
, Runzhi Chen 3,*1 College of Life Science and Technology, Huazhong Agricultural University, 430070 Wuhan, Hubei, China
2 College of Biomedicine and Health, Huazhong Agricultural University, 430070 Wuhan, Hubei, China
3 Department of Medical Oncology, Huazhong University of Science and Technology, Tongji Medical College, Hubei Cancer Hospital, 430079 Wuhan, Hubei, China
†These authors contributed equally.
Abstract
Choline participates in three major metabolic pathways: oxidation, phosphorylation, and acetylation. Through oxidation, choline is converted to betaine and contributes to methyl metabolism and epigenetic regulation. Through phosphorylation, choline participates in phospholipid metabolism, and serves as the precursor of phosphocholine, phosphatidylcholine, glycerophosphocholine, and other essential compounds, thereby modulating lipid metabolism and transport. Through acetylation, choline is transformed into acetylcholine in cholinergic neurons, playing a vital role in neurotransmission. Moreover, gut microbiota can metabolize choline into trimethylamine-N-oxide, and be involved in the pathogenesis of various diseases such as nonalcoholic fatty liver disease (NAFLD), cancer, cardiovascular disease, etc. Since choline metabolism is implicated in the development of NAFLD and diverse cancers, including liver cancer, it may serve as a therapeutic target for these diseases in the future. Currently, there are numerous therapeutic agents targeting choline metabolism to treat NAFLD and cancers, but most of them are ineffective and some even have adverse effects that lead to a series of complications. Therefore, further research and clinical validation are required to obtain safe and efficacious drugs. This review comprehensively summarizes the choline metabolic pathway and its regulatory mechanisms, elucidates the roles and mechanisms of choline metabolism in the aforementioned diseases, and provides a discussion of the current advances and immense potential of this field.
Keywords
- choline
- acetylcholine
- betaine
- phosphatidylcholine
- nonalcoholic fatty liver disease
- cancer
Choline, a nutrient procured both through dietary intake and endogenous synthesis, is fundamentally a methyl-rich quaternary amine, which manifests in all mammalian tissues either freely or in an esterified form [1]. In 1998, the Institute of Medicine acknowledged choline as an essential nutrient and proposed pertinent dietary guidelines [2]. Noteworthy sources of dietary choline include animal liver, eggs, and wheat germ, and it exists freely in food and is also found in the enzyme cholinesterase [3]. Choline participates in lipid transport, methylation metabolism, cell membrane signal transduction, and neurotransmitter synthesis through oxidation, phosphorylation, and acetylation pathways [4].
Studies have demonstrated that low choline intake (approximately
The polymorphism of enzymes in choline metabolic pathway may be related to the occurrence of cancer. A case-control study in China revealed a negative correlation between dietary choline intake and the risk of breast cancer. The polymorphism of phosphatidylethanolamine-N-methyltransferase (PEMT) rs7946 and betaine homocysteine methyltransferase (BHMT) rs3733890 may be related to the effect of choline intake on breast cancer risk. Low choline intake was associated with a significantly increased risk of breast cancer in women with wild-type PEMT rs7946 or BHMT rs3733890 [7]. Moreover, the increased resonance of total choline compounds (tCho) in vivo has also been reported as a common feature of many cancers [8]. The changes of choline phospholipid metabolism detected by non-invasive magnetic resonance spectroscopy can be used as endogenous tumor biomarkers. Similarly, choline kinase, phosphatidylcholine specific phospholipase C and D, and other choline metabolism related enzymes are considered as new targets of tumor therapy [4, 9].
Choline also promotes cell growth and division [10]. Some studies have shown that choline and dimethylglycine produce superoxide/hydrogen peroxide through the electron transport chain of liver mitochondria. Hydrogen peroxide is a mitotic factor and a mitochondrial signal molecule, which coordinates mitochondrial metabolism and cell physiological changes. The essential metabolites involved in the production of methionine and folate are the sources of Reactive Oxygen Species (ROS) in liver mitochondria. ROS signal transduction from mitochondria to the rest of cells is involved in regulating various functions of cells, including division, growth, and many other functions [11].
Choline, is a trimethyl compound. In the liver and kidney, choline oxidase system (composed of choline dehydrogenase and betaine aldehyde dehydrogenase) first catalyzes the formation of betaine aldehyde and then converts it into betaine [7, 12, 13]. Betaine is an osmotic regulator that can be accumulated or released according to the cell volume and plays an important role in the homeostasis of human body [4, 12], as well as in protecting the liver [13]. It has been reported that betaine supplementation can increase plasma low density lipoprotein (LDL) cholesterol and reduce plasma high density lipoprotein (HDL) cholesterol [14]. A meta-analysis showed that high betaine levels were associated with a lower risk of cancer, particularly colorectal cancer [14]. According to another report, betaine intake may be inversely proportional to the risk of fatal prostate cancer in white and African American men [15]. In addition, choline and betaine are involved in neurodevelopment and the pathogenesis of various chronic diseases, playing an important role in risk assessment and disease prevention [4].
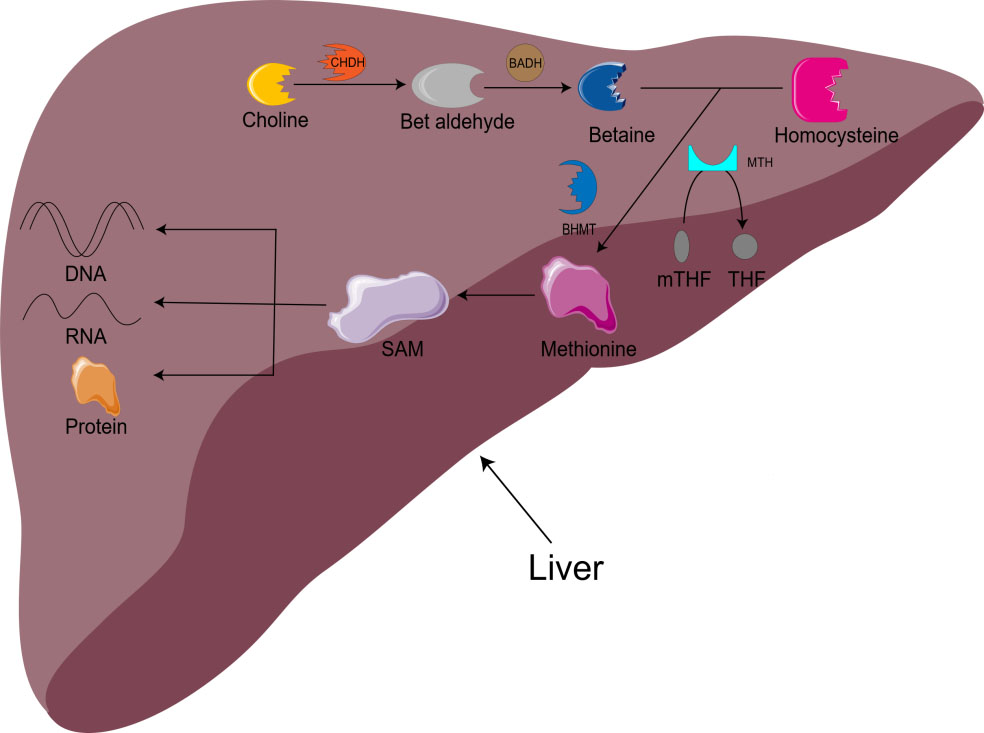
Choline is also involved in methyl metabolism. In the liver, choline is irreversibly oxidized to betaine. With the assistance of BHMT and methionine synthetase (MTH), homocysteine is remethylated to methionine to produce S-adenosylmethionine (SAM) [16]. SAM is the main biological methylation agent, responsible for the methylation of DNA, RNA, and protein, as well as the biosynthesis of small molecules such as creatine, adrenaline, and phosphatidylcholine [1, 5, 11]. 1-carbon metabolism is essential for metabolism, cell proliferation, nerve function, signal transduction, and tissue development, and can be used in purine, amino acid and thymine biosynthesis (Fig. 1) [11]. Animal model studies have shown that there is an important relationship between choline/1-carbon metabolism and energy homeostasis [5].
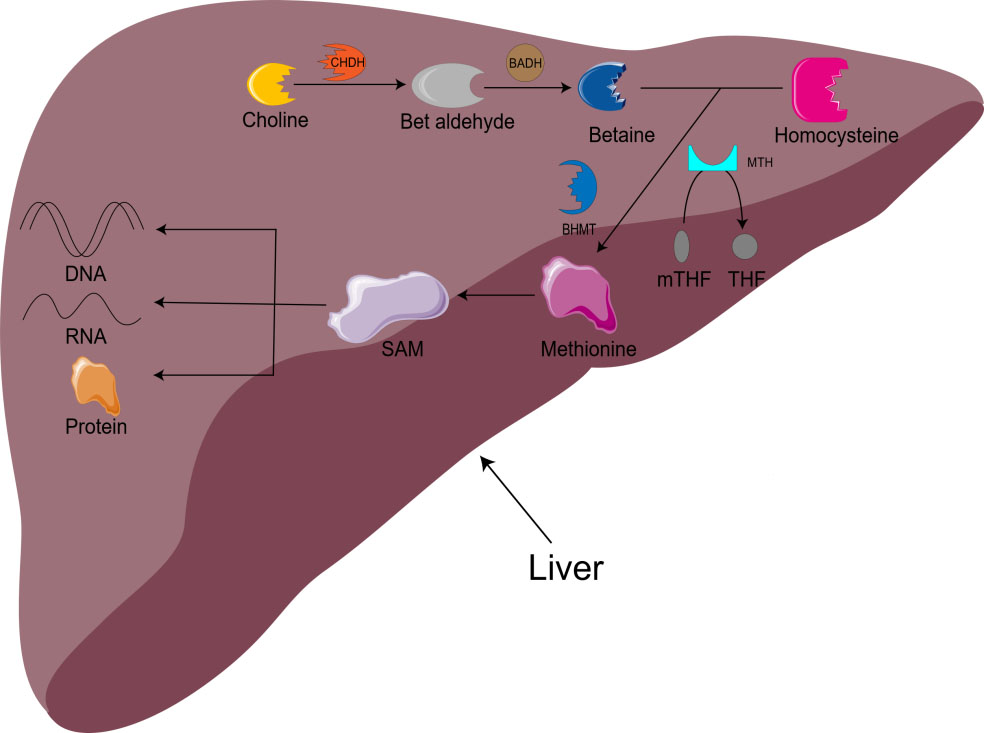 Fig. 1.
Fig. 1.Metabolism of choline-betaine. CHDH, choline dehydrogenase; BADH, betaine aldehyde dehydrogenase; mTHF, 5-methyltetrahydrofolate; THF, Tetrahydrofolate; BHMT, betaine homocysteine methyltransferase; MTH, methionine synthetase; SAM, S-adenosylmethionine.
Phospholipid metabolism is critical for cell proliferation as it manufactures the components required in the cell membrane and intracellular signaling. Mammalian cells synthesize phosphatidylcholine and phosphatidylethanolamine, the most abundant phospholipids in the cell membrane, through the Kennedy’s pathway and integrate them into the membrane [12, 17]. The synthesis of phosphatidylcholine and phosphatidylethanolamine are called Cytidine Diphosphate Choline (CDP-choline) and CDP-ethanolamine pathways respectively [9].
Phosphatidylcholine (PC) is found extensively on mammalian cell membrane and acts as an important source of intermediate products of intracellular signal transduction pathway (such as second messenger phosphocholine, diacylglycerol and arachidonic acid metabolites, which are essential for cell mitotic activity) [18]. Inhibition of the synthesis of phosphatidylcholine in mammalian cells (including cancer cells) has been proved to induce cell death [19, 20]. Phosphatidylcholine is also the most important source of homocysteine in mammalian tissues, accounting for 95% of the total choline pool in mammalian tissues. The remaining 5% includes choline, phosphocholine, glycerophosphocholine, cytidine 5-diphosphate choline, and acetylcholine [4, 21].
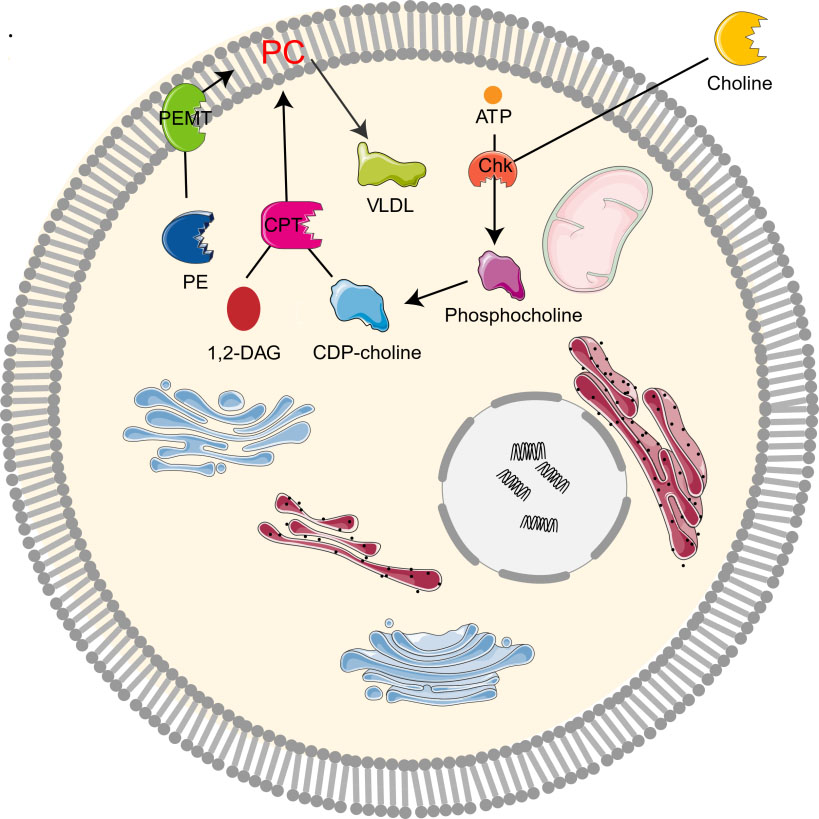
The study on the synthesis and metabolism of phosphatidylcholine in mice showed that 70% of the synthesis of liver phosphatidylcholine came from the CDP-choline pathway while the other 30% came from the PEMT pathway [1, 2, 10]. As the CDP-choline pathway is essential for providing cell membrane and lipid secondary messengers, it is a key determinant of cell cycle progression, cell proliferation, and programmed cell death (apoptosis) [10]. Choline kinase (Chk) is a kind of cytosolic enzyme that exists in many different types of tissues. It catalyzes the phosphorylation of choline to form phosphocholine in the presence of Adenosine triphosphate (ATP) and magnesium [22]. In the CDP-choline pathway, Choline enters cells through the action of transporters by crossing biological membranes, then choline kinase catalyzes the phosphorylation of choline by ATP hydrolysis to produce choline phosphate, which is then modified to cytidine diphosphate (CDP)-choline, and CDP-choline: 1,2-diacylglycerol cholinesterase (CPT) catalyzes the conversion of CDP-choline and diacylglycerol to phosphatidylcholine [12, 19]. Phosphocholine is both the precursor and the decomposition product of phosphatidylcholine. Together with other phospholipids such as phosphatidylethanolamine and neutral lipids, phosphatidylcholine forms a unique double-layer structure in the cell membrane and regulates the integrity of membrane [9]. In addition, PEMT pathway refers to the de novo synthesis of PC from phosphatidylethanolamine (PE) by the S-adenosylmethionine (SAM, intermediate product of methionine cycle) dependent enzyme PEMT [4, 10, 23]. Moreover, PC can be degraded by many phospholipases to form lipid intermediates, which can then be recycled or further processed [24]. It can also be decomposed by phospholipase A2 (PLA2), C, D, and phosphatidylserine synthetase 1, or converted to sphingomyelin (Fig. 2).
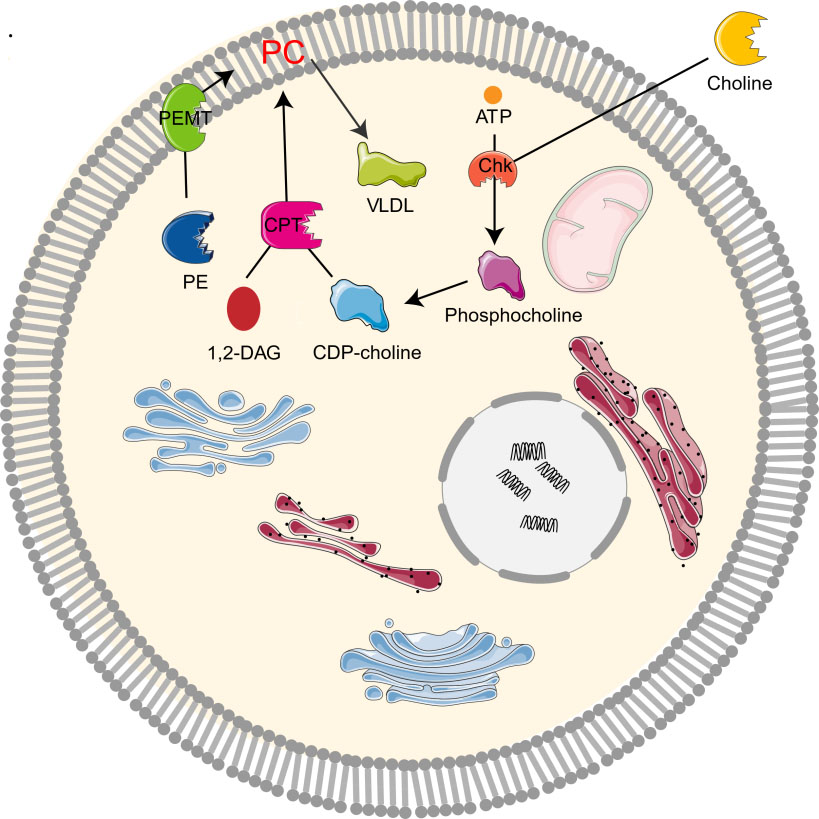 Fig. 2.
Fig. 2.Metabolism of choline-phosphatidylcholine. Chk, choline kinase; CDP-choline, cytidine diphosphate-choline; 1,2-DAG, 1,2-diacylglycerol; CPT, 1,2-diacylglycerol cholinephosphotransferase; PE, phosphatidylethanolamine; PC, phosphatidylcholine; PEMT, phosphatidylethanolamine N-methyltransferase; VLDL, very low density lipoprotein.
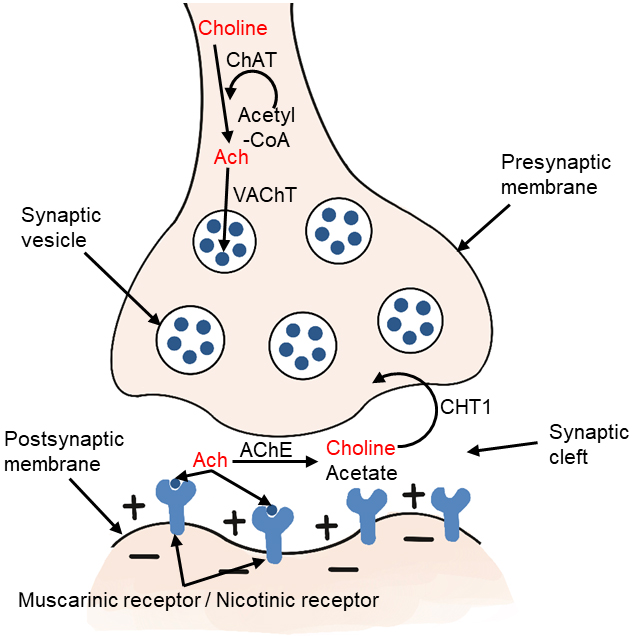
Choline-acetyltransferase (ChAT) is an enzyme highly concentrated in cholinergic nerve terminals. It catalyzes the reaction of acetyl-CoA with choline to produce acetylcholine, which acts as a neurotransmitter [2, 12, 25]. The ChAT in presynaptic cholinergic neurons synthesize acetylcholine, which is then absorbed into synaptic vesicles by vesicular acetylcholine transporter (VAChT). After depolarization, acetylcholine is excreted. When it reaches the synaptic cleft, it binds to the receptors of postsynaptic neurons in the central and peripheral nervous systems. There are two types of receptors, including muscarinic and nicotinic receptors. Acetylcholine in the synaptic cleft is rapidly hydrolyzed by acetylcholinesterase (AChE) to form acetate and choline, which are circulated into presynaptic nerve endings by high affinity choline transporter (CHT1) (Fig. 3). This reuptake through the transporter contributes to the formation of the choline pool, which will be used by cholinergic neurons for the resynthesis of acetylcholine or the formation of phosphatidylcholine back to the biomembrane. Cholinergic neurons in the basal forebrain, including those forming the Meynert basal nucleus, are severely lost in Alzheimer’s disease (AD) [25, 26]. Acetylcholine synthesis has also been reported in tissues, including placenta, muscle, intestine, and lymphocytes [2].
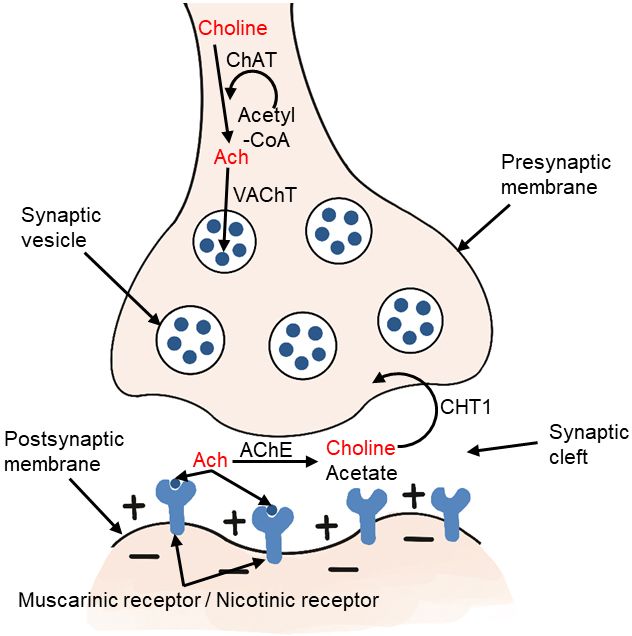 Fig. 3.
Fig. 3.Metabolism of choline-acetylcholine in cholinergic neurons. ChAT, choline acetyltransferase; Ach, acetylcholine; VAChT, vesicular acetylcholine transporter; AChE, acetylcholinesterase; CHT1, high affinity choline transporter.
NAFLD is the most common liver disease in adults and children in the world. The pathophysiology of NAFLD is multifactorial, stemming from a complex interplay of ecological, genetic, and metabolic factors, such as high energy intake, limited physical activity, and malnutrition [27]. Combined with epigenetic factors, these promote insulin resistance and liver fat accumulation [13, 28].
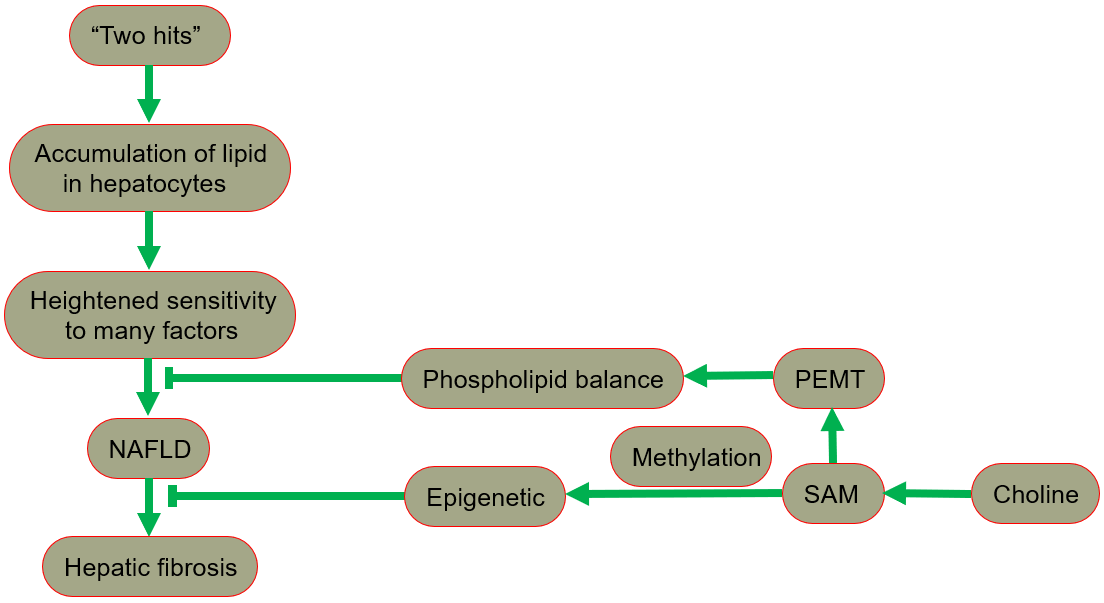
The hypothesis of “two hits” is proposed for the development of NAFLD [29, 30], in which “the first hit” involves the accumulation of lipid in hepatocytes. In fact, the increase in lipid uptake, the increase in de novo synthesis of fat, the decrease in lipoprotein synthesis, or the decrease in fatty acid oxidation may all lead to lipid accumulation. After the “first hit”, the liver has a heightened sensitivity to many previously inconsequential factors, including gut derived endotoxin, endoplasmic reticulum (ER) stress, oxidative stress and subsequent lipid peroxidation, pro-inflammatory cytokines and adipocytokines signals, and mitochondrial dysfunction. These effects result in the “second hit”, which promotes liver cell damage and death, inflammation, activation of hepatic stellate cells (HSCs), and production of extracellular matrix, eventually leading to fibrosis [30, 31]. A fatty liver could develop into liver damage accompanied by fibrosis. The process of fibrosis begins with the activation of HSCs by cytokines and growth factors, while the subsequent acquisition of a new set of epigenetic markers maintains this new cell phenotype. Low levels of inflammatory signaling combined with epigenetic mechanisms usually keep HSCs latent, so a reduction in the availability of methyl donors can induce and maintain hepatic fibrosis [32].
Phospholipid balance, paramount for liver health, is maintained primarily by PEMT [33, 34, 35]. In the presence of PEMT, endogenous synthesis of phosphatidylcholine is possible, reducing the reliance on dietary choline [13]. When choline intake is insufficient, the PEMT pathway that synthesizes phosphatidylcholine becomes very important for maintaining the supply of phosphatidylcholine in the liver. Since the pathway occurs only in the presence of SAM, the regulation of SAM is essential in ensuring a sufficient phosphatidylcholine supply [1].
Any malfunctions in the metabolism of choline phospholipid could lead to hepatic steatosis, which can induce fatty liver. Hepatic steatosis occurs when fatty acid intake, de novo synthesis of fatty acid, or triglyceride synthesis in the liver exceeds the output or oxidation of lipids [1, 36]. Phosphatidylcholine is necessary for the assembly, secretion, and dissolution of cholesterol in bile. The homeostasis of phosphatidylcholine is achieved by balancing the uptake/synthesis of phosphatidylcholine, choline, and betaine, as well as the catabolism/secretion of phosphatidylcholine [1]. Phosphatidylcholine deficiency increases the de novo synthesis of saturated fatty acid in the liver. Compared with people with healthy liver, NAFLD patients exhibit an excessive level of triglycerides rich in saturated fatty acids, along with an increased risk of fatty liver disease, pancreatitis, cirrhosis, and liver cancer. This indicates that the increase in de novo synthesis of fat will lead to hepatic steatosis [1, 23]. Phosphatidylcholine is also involved in the assembly and secretion of very low density lipoprotein (VLDL) [4, 37]. It is necessary for the packaging and export of triglycerides in VLDL and the dissolution of secretory bile salts. VLDL granules transport fat and cholesterol from the liver to peripheral organs. Abnormal VLDL mediated triglyceride secretion is the main culprit of hepatic steatosis [13, 37, 38]. The assembly and secretion of liver VLDL is a complex process, involving apolipoprotein B (ApoB), microsomal triglyceride transfer protein (MTP), lipid mobilization, and synthesizing proteins. In fact, rare ApoB and MTP mutations are associated with progressive liver disease [23, 39].
The occurrence and development of NAFLD are closely related to obesity and
insulin resistance. The accumulation of visceral adipose tissue is the main
reason for an increase in fatty acids, proinflammatory regulators, and growth
promoting regulators, it is closely related to the progress of nonalcoholic
steatohepatitis (NASH) [40]. A low choline diet can lead to fatty liver and liver
injury, possible due to the accumulation of fat and cholesterol in liver [4, 37].
This dietary requirement for choline is regulated by estrogen and single
nucleotide polymorphisms (SNPs) in specific genes in choline and folate
metabolism [3]. The classical function of estrogen is realized by its receptor
ER
Lipid homeostasis in the liver strikes a fine balance between uptake and
synthesis, as well as secretion and catabolism. This homeostatic equilibrium is
disturbed in Pemt
Chronic impairment of lipid metabolism affects the balance of
oxidants/antioxidants, influencing the functionality of metabolism-related
organelles, leading to chronic endoplasmic reticulum stress (ERS), and
mitochondrial dysfunction. The interaction between ERS and oxidative stress plays
a crucial role in the pathogenesis of NAFLD [42]. Disruption of
mitochondria-associated membrane (MAM) integrity results in an imbalance in
Ca
Hepatocytes are subjected to sustained stress or injury, and the activation of macrophages, leading to activation of resident hepatic stellate cells into myofibroblasts, which resulting in the accumulation of extracellular matrix. This process ultimately leads to fibrosis, cirrhosis, and even progression to hepatocellular carcinoma [47]. Low dietary choline result in steatohepatitis, fibrosis, cirrhosis. Studies have shown that Exosomes derived from human adipose mesenchymal stem cells (hADMSCs-Exo) can promote choline uptake, reduce lipid accumulation, and restore abnormal choline metabolism, thereby helping to alleviate liver cell damage and fibrosis. On this basis, choline supplementation is supposed to exert a synergistic effect with hADMSCs-Exo in anti-hepatic fibrosis [48].
In summary, NAFLD includes a series of pathologies, ranging from nonalcoholic fatty liver (NAFL, commonly referred to as simple steatosis, defined as steatosis without any histological or biochemical damage) to NASH, characterized by steatosis, inflammation and hepatocyte damage (swelling), with or without cirrhosis. NAFL is generally considered as a benign disease, and NASH is a progressive disease. Patients with NASH are likely to develop advanced fibrosis and cirrhosis, or even liver cancer (Fig. 4) [28, 49, 50].
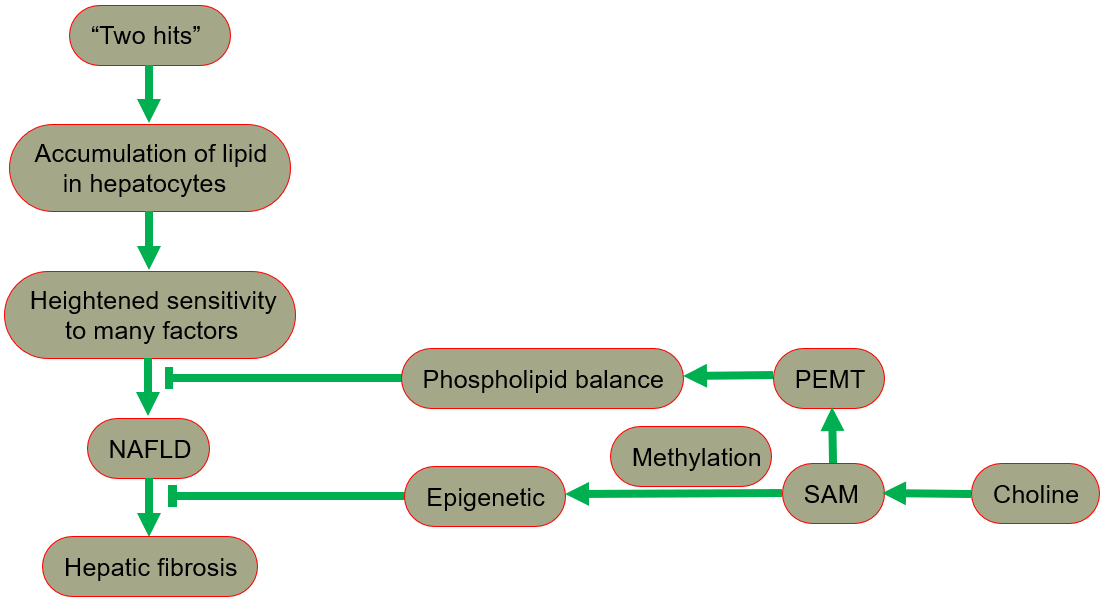 Fig. 4.
Fig. 4.The protective role of choline in the development of NAFLD. NAFLD, nonalcoholic fatty liver disease; PEMT, phosphatidylethanolamine-N-methyltransferase; SAM, S-adenosylmethionine.
The gut microbiota is mainly composed of bacteria, viruses, fungi, etc. Although gut microbiota is individual specific and has high adaptability to change, it could still be affected by the host’s internal and external factors, such as the host’s genes, dietary habits, presence of antibiotics, and environmental changes [51]. An increasing library of data shows that certain diseases, such as irritable bowel syndrome, inflammatory bowel disease, diabetes, allergy, cancer, obesity, autism, and liver disease are related to the gut microbiota dysregulation [52]. It has been reported that the liver steatosis phenotype is affected by gut microbiota [13, 28]. Gut microbiota can produce a variety of compounds, which play an important role in regulating the activities of distal organs, while the liver is in a strategic position downstream of the intestine. The anatomy of the liver ensures a close connection with the intestine, allowing enterogenous bacteria and their metabolites, nutrients, and other signals to be transmitted to the liver through the portal vein circulation. The liver also plays a crucial role in the defense of enterogenous substances (this is defined as the gut-liver axis). Gut microbiota, as a bioreactor, is equipped with the ability to engage in autonomous metabolism and immunity. It can regulate the response to external stimuli in the host environment, and the complexity of gut microbiota can indicate that it is an organ. Therefore, the concept of gut-liver axis must be complemented by gut-microbial-liver network, because of the complexity of microbial components and metabolic activities. Compounds related to gut microbiota, such as short chain fatty acids, bile acids, choline metabolites, indole derivatives, vitamins, polyamines, lipids, neurotransmitters and neuroactive compounds, and hypothalamus-pituitary-adrenal axis hormones have a variety of biological functions [29, 52].
Gut microbiota plays an important role in the pathophysiological process of cirrhosis. Enterogenous complications of cirrhosis, such as small intestinal bacterial overgrowth (SIBO) and increased intestinal permeability (intestinal leakage), can lead to systemic diseases related to bacterial or endotoxin translocation, such as spontaneous bacterial peritonitis, portal hypertension, hepatorenal syndrome, hepatic encephalopathy, and multiple organ failure [52]. SIBO refers to an increase in the number of anaerobes and/or aerobic bacteria in the small intestine, combined with the increase in intestinal permeability. SIBO is considered to be one of the pathogenesis of NAFLD/NASH. The intestinal epithelium is a natural barrier to prevent harmful bacteria and elements from entering the circulation, but the translocated microorganisms in SIBO may damage the tight connection between small intestinal epithelial cells, increase intestinal permeability, promote the flow of bacterial byproducts in the portal system, and absorb them from the liver, showing toxic activities in the liver. SIBO also induced the expression of Toll-like receptor 4 (TLR4) and the release of interleukin-8 (IL-8), which stimulated the inflammatory response [29, 53, 54].
Data from preclinical and clinical studies indicate that gut microbiota plays a key role in the pathogenesis of NAFLD, mainly through obesity tendency, metabolic changes (insulin resistance) and promoting liver inflammation [29, 45, 53]. In addition, many byproducts of bacteria, such as ethanol, may exhibit hepatotoxicity by stimulating Kupffer cells to produce and secrete nitric acid and cytokines [29, 51].
Obesity is a common feature of most metabolic diseases. Gut microbiota also plays a key role in the occurrence and development of obesity and obesity related metabolic diseases through the production of short chain fatty acids and other microbial metabolites that can regulate the energy gain of the host, or through the signal pathway that regulates the energy metabolism of the host. Studies have shown that gut microbiota can promote the absorption of monosaccharides in the intestine, accelerate the de novo synthesis of liver fat, inhibit the adipocytokines induced by fasting, and lead to the accumulation of triglycerides in adipocytes [53].
Insulin resistance is the basic pathophysiological process of metabolic diseases. In NAFLD, insulin resistance accelerates fatty accumulation in hepatocytes, leading to hepatocyte injury and inflammation. Inflammation and increased insulin resistance form a “vicious circle” that worsens the development of NAFLD [53, 54].
The term “metabolic endotoxemia” refers to a metabolic disease where there is an increased level of lipopolysaccharide (LPS) in the blood. LPS is the active component of endotoxin, a part of the cell membrane of Gram-negative bacteria, and can bind to LPS binding protein and monocyte differentiation antigen CD14 receptor to form a complex that interacts with Toll-like receptors (TLRs) and activates inflammatory cascade reaction [29, 55]. Since both NAFLD and NASH are characterized by increased endotoxin levels, endotoxin is recognized as one component of the “second hit” in the “two hits” hypothesis [56, 57], triggering a cascade of pro-inflammatory cytokines, insulin resistance and triglyceride production, thus promoting the occurrence and development of NAFLD [29].
Any changes in gut microbiota, especially that of E. coli and other Enterobacteriaceae, will lead to gut microbiota dysregulation and increase the production of endogenous ethanol [51]. The increase in ethanol levels in obese patients and nonalcoholic NASH children suggests its role in the development of NAFLD/NASH. In addition, the expression of ethanol metabolism related enzymes such as ethanol dehydrogenase, catalase, and aldehyde dehydrogenase in NASH liver was significantly increased [29]. In addition to promoting inflammation, ethanol can also promote oxidative damage and hepatocyte necrosis because it can form reactive oxygen species and nitrogen. Ethanol can also increase the activity of the cytochrome P450 2E1 (CYP2E1) enzyme, which catalyzes the oxidation of ethanol and releases free radicals, leading to oxidative damage, mitochondrial dysfunction, and liver inflammation. Endogenous ethanol can also inhibit the tricarboxylic acid cycle, thus increasing the level of acetic acid, thus promoting the accumulation of triglycerides in hepatocytes [51].
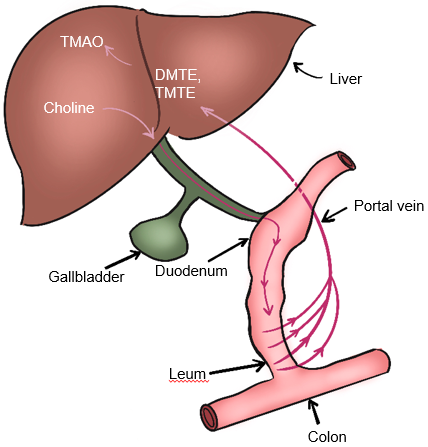
Gut microbiota also participate in choline metabolism by converting choline into toxic dimethylamine and trimethylamine. As shown in Fig. 5, dimethylamine and trimethylamine are transported to the liver and then oxidized by monooxygenase containing hepatoflavin to transform them into Trimetlylamine oxide (TMAO)), which leads to liver inflammation and damage [2, 53, 58, 59]. The increase of TMAO production is also the main cause of cardiovascular disease. Studies have shown that there is a positive correlation between circulating TMAO level and the occurrence and severity of NAFLD in Chinese adults in hospitals and communities [60]. Many studies have shown that TMAO is a gut microbiome-dependent metabolite of choline, which can lead to the occurrence and development of cardiovascular diseases [52, 60]. It has also been reported that TMAO can regulate glucose and lipid metabolism by reducing the total bile acid pool size, affecting lipid absorption and cholesterol homeostasis [60]. In addition, the content of dietary choline affects the composition and abundance of gut microbiota related to NAFLD [53]. The close relationship between gut microbiota and choline metabolism provides an important theoretical basis for targeted therapy of gut microbiota of NAFLD.
 Fig. 5.
Fig. 5.Metabolism of choline-TMAO. DMTE, dimethylamine; TMTE, trimethylamine; TMAO, Trimethylamine-N-oxidate.
NAFLD patients are at an increased risk of death from cardiovascular
disease and diabetes, so there is a growing need for new and better diagnostic
methods and treatments [29, 61]. Peroxisome proliferator-activated receptors
(PPARs) are members of the nuclear hormone receptor superfamily, which can
directly regulate target gene transcription by forming a heterodimer with the
retinoid receptor X (RXR) and binding to the PPAR response element (PPRE)
sequences in gene promoters [62]. There are three subtypes of PPAR:
| Types | Drugs | Therapeutic effect | References |
| PPAR |
Fenofibrate | Improved liver histology and biomarkers in NASH. | [41] |
| Pemafibrate | Reduced based liver stiffness in NASH and improved serum alanine aminotransferase (ALT). | [67] | |
| PPAR |
Pioglitazone | Well-tolerated and effective in improving liver histology and reducing liver steatosis. | [68, 69] |
| Rosiglitazone | Significantly decreased liver steatosis. | [70] | |
| Lobeglitazone | Reduced intrahepatic fat content, improved glycemic, liver, and lipid profiles in NAFLD. | [71] | |
| PPAR |
Elafibranor | Improves insulin sensitivity, glucose homeostasis, and lipid metabolism and reduces inflammation. | [72] |
| PPAR |
Saroglitazar | Significantly improved ALT, insulin resistance, and atherogenic dyslipidemia in NAFLD/NASH. | [73] |
| PPAR |
Lanifibranor | Simultaneously improved insulin resistance and improved the fibrotic response, and reduced hepatic inflammation in NASH. | [72, 74] |
PPAR, Peroxisome proliferator-activated receptor; NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis.
PPAR
PPAR
PPAR
Although PPAR
Farnesoid X receptor (FXR) is another kind of nuclear receptor and highly
expressed in both hepatocytes and intestinal enterocytes where it controls all
aspects of bile acid metabolism. It is endogenously activated by bile acids,
which can improve insulin sensitivity and show direct anti-inflammatory and
anti-fibrosis effects in NASH mouse model and human tissues [80]. These compounds
are FXR agonists, whose prototype is Obeticholic Acid (OCA), which can improve
insulin resistance [81]. The GLS1-subtype of glutaminase was enhanced in both
NASH clinical biopsy and preclinical mouse models. Since GLS1 silencing
significantly reduced steatosis and oxidative stress through complex metabolic
reprogramming (including increased VLDL output), this suggests that GLS1 may be
an effective target for the treatment of NASH [23]. Recently, a benzimidazole
derivative has been designed that possess FXR/PPAR
Since gut microbiota plays a key role in the occurrence and development of NAFLD, gut microbiota has the potential to be the target for the treatment of NAFLD. Probiotics, prebiotics, and synbiotics have demonstrated their therapeutic potential in modulating the microbiota composition and promoting microbial equilibrium [83]. Recent studies showed that gut microbiota also can be regulated by antibiotics such as Vancomycin [84] and some Traditional Chinese Medicine (TCM) formulas such as Qushi Huayu Decoction (QHD) [85].
Probiotics was defined as “living microorganism” by the World Health
Organization in 2001. Bifidobacterium and Lactobacillus are the
main probiotics, also called traditional probiotics, which are considered to be
harmless to the human body [52, 53]. Only in recent times have researchers begun
to comprehend the mechanisms of their functionality by employing rodent models.
In animal models with diet-induced NAFLD, the administration of oral probiotics
containing Lactobacillus fermentum has demonstrated the ability to
decrease hepatic lipid accumulation, oxidative stress, and the expression of
inflammatory markers (such as Tumor necrosis factor-
| Probiotic strains | Treatment results | References |
| A. muciniphila | Reversed HFD-induced metabolic disturbances, improved lipid metabolism, and epigenetic regulation. | [92, 93] |
| B. uniformis | Reduced liver steatosis, serum cholestero and triglyceride, and increased small adipocyte numbers. | [94, 95] |
| B. xylanisolvens | Delayed progression of NAFLD to nonalcoholic steatohepatitis and improved nicotine-associated NAFLD. | [96] |
| E. hallii | Improved insulin sensitivity, increased energy expenditure, improved metabolic phenotype. | [97] |
| F. prausnitzii | Improved hepatic health, decreased adipose tissue inflammation, enhanced mitochondrial respiration. | [98] |
| Propionibacterium | Reduced significantly TNF- |
[99] |
| Clostridia | Helped T-cell expansion and differentiation, and reduced intestinal inflammation in adult mice. | [100] |
NGPs, novel generation of probiotics; HFD, high-glucose diet; TNF-
In addition, synergistic effects have been observed in the combination of probiotics and chemicals such as metformin in NASH treatment and statins in NAFLD treatment, which highlights the great potential of probiotics alone or in combination with other drugs [71]. However, the clinical efficacy of probiotics still needs to be further validated in well-designed and larger-scale studies.
Prebiotics are a class of non-digestible food ingredients that selectively alter the growth and/or activity of bacteria in the colon [102]. They selectively stimulate “good” bacteria in the colon and inhibit the growth and/or activity of “bad” bacteria [103]. Oligosaccharides such as fructans and galactans are as examples of prebiotics for humans, which promote the proliferation of Bifidobacteria [103]. There is evidence that prebiotic supplements can prevent the development of NAFLD in both experimental and clinical studies. The combination of prebiotics and natural ingredients will produce greater benefits than prebiotics themselves [30]. For example, Isomaltooligosaccharides (IMOs) combined with lycopene (an antioxidant) can prevent high-fat diet induced weight gain in NAFLD mice, enhance fat mobilization in adipose tissue, and improve insulin resistance and metabolic endotoxemia [52].
The combination of probiotics and prebiotics is called synbiotics [29, 52, 53]. Synbiotics usually benefit by selectively stimulating the growth of healthy bacteria and/or activating their metabolism. Synbiotics have shown a variety of benefits in metabolic diseases, such as improving insulin resistance, blood glucose control and inflammatory cytokine synthesis. Synbiotic therapy showed improvements in fasting blood glucose, triglycerides, and inflammatory cytokines in obese and lean NAFLD patients. Therefore, synbiotics is a promising method for the prevention or treatment of NAFLD targeted by gut microbiota. However, more clinical validation is needed [53].
Antibiotics are often used clinically, although their ability to disrupt the homeostasis of gut microbes is a double-edged sword. On the one hand, the short-term use of antibiotics will have a long-term impact on host metabolism; on the other hand, the use of some antibiotics can improve the disease. Compared with antibiotics, some components of Chinese herbal medicine have a greater prospect of regulating gut microbiota with less side effects. Berberine is a typical component of Chinese herbal medicine, which has strong antibacterial activity, especially for intestinal bacteria, because berberine is difficult to be absorbed in the intestinal tract. At present, more and more evidences confirm that berberine has therapeutic effects on metabolic diseases, including obesity, NAFLD, and type Ⅱ diabetes (T2D), by regulating gut microbiota [53].
Choline and betaine can be metabolized by gut microbiota to form trimethylamine (TMA), which is further oxidized to trimethylamine N-oxide (TMAO) in the liver. Studies have found that TMAO can enhance the anti-tumor immune response in pancreatic cancer. Intraperitoneal administration of TMAO or oral supplementation of choline can suppress Pancreatic ductal adenocarcinoma (PDAC) growth, by activating tumor-associated macrophages (TAM) and effector T cells, and promoting the type I interferon (IFN) pathway in the tumor microenvironment. Combining TMAO with Immune Checkpoint Blockade (ICB) in a PDAC mouse model significantly reduced tumor burden. This indicates that the gut microbiota metabolic product TMAO can act as a driving factor for anti-tumor immunity and lays the foundation for potential therapeutic strategies targeting TMAO [104]. However, in colon cancer, it has been found that TMAO promotes tumor growth by upregulating vascular endothelial growth factor A (VEGFA) expression [105].
With the rapid development of immune therapy in tumors, it has been found that
tumor metabolism plays an important role in immune suppression and the immune
microenvironment [106, 107]. Bioinformatics analysis revealed that two
metabolism-related genes, choline kinase beta (CHKB) and Phosphatase and tensin
homolog (PTEN), were upregulated in colon cancer. The high-risk group had worse
prognosis, and there was a significant difference in immune cell distribution
between the two groups, with increased expression of CD8
In addition, Tumor microenvironment (TME) can affect choline metabolism [9]. TME has complex components, including all kinds of non-tumor cells and non-cellular substances. Immune cells are the key components of TME. Metabolites in TME, such as fatty acids and glucose, can regulate the metabolism, phenotype and function of immune cells. These affected immune cells can, in turn, significantly affect the adjacent tumor cells [112]. Studies have shown that 27-hydroxycholesterol, a cholesterol metabolite, promotes breast cancer metastasis by acting on immune cells [56]. Metabolic reprogramming is necessary for tumor cells to maintain growth and survival in adverse microenvironment. In a study on choline metabolism of breast epithelial cells cultured in vitro, it was found, for the first time, that the total amount of phospholipid metabolites containing choline increased with the transformation from normal cells to tumor cells. In addition, the cultured malignant breast epithelial cells absorbed and/or metabolized more choline than the normal cells with the same growth rate. It can be inferred that the malignant transformation of breast tissue is related to abnormal choline metabolism [57].
The molecular mechanism of abnormal choline metabolism is the change of choline
kinase-
As a polar compound, choline can only enter cells through transporters on the cell membrane. Many transporter systems and enzyme networks are involved in phosphatidylcholine metabolism, but they are often dysregulated in cancer cells. Increased transport of extracellular free choline into cancer cells is considered a major cause of choline phenotype changes in cancer cells, as this may be the limiting step in phosphatidylcholine formation in some cases. There are four different types of choline transporters related to cancer, classified according to their affinity: (a) high-affinity choline transporters (CHTs); (b) choline transporter-like proteins (CTLs); (c) organic cation transporter (OCTs); (d) organic cation/carnitine transporters (OCTNs). The expression levels of each transporter subtype are increased in cancer cells [9, 116]. Choline transport mediated by CTL1 is considered a fundamental function in phospholipid synthesis, and inhibiting this choline uptake function can induce cell apoptosis. CTL1 is upregulated in various malignant tumors, including central nervous system, breast cancer, prostate cancer, melanoma, kidney cancer, lung cancer, colon cancer, and pancreatic cancer. High expression of CTL1 and CTL2 mRNA was observed in the MIA PaCa-2 pancreatic cancer cell line, and inhibiting CTL1 function resulted in cell apoptosis. CTL1 inhibitors Amb4269951 and Amb4269675 significantly inhibited tumor growth in a mouse xenograft model, indicating that CTL1 is a target molecule for the treatment of pancreatic cancer [117]. Human lung adenocarcinoma tissue highly overexpresses CTL1, and partially overexpresses organic cation transporters 3 (OCT3). In the NCI-H69 cell line, the increase in choline uptake depends mainly on CTL1, partially on OCT3, OCTN1, and OCTN2 [118].
Choline kinase (Chk) is the first enzyme in the choline metabolism pathway,
which converts choline to PC. Chk was first discovered in 1953. There are three
subtypes of ChK in mammalian cells, Chk-
Tumor development and metastasis involve epithelial mesenchymal transition (EMT). Some studies have shown that inhibition of phosphatidylcholine specific phospholipase C (PC-PLC) can lead to the loss of mesenchymal properties of metastatic breast cancer cells, indicating that PC-PLC plays an important role in the molecular pathway of maintaining mesenchymal like phenotype of metastatic breast cancer cells. Inactivation of PC-PLC may be a means to promote the differentiation of breast cancer cells and improve the effect of anti-tumor therapy [68]. Phospholipase D (PLD) was first discovered in plants in 1947 [123]. PLD is an enzyme belonging to the phospholipase superfamily, and the main subtypes in human cells are PLD1 and PLD2. PLD can hydrolyze phosphatidylcholine into phosphatidic acid and choline. The activity of PLD is significantly increased in tumor tissues and cells, including colorectal cancer, breast cancer, stomach cancer, thyroid cancer, brain cancer, and renal cancer, indicating that it may play a key role in the progression of cancer [124].
In addition, the promoter sequence of Chk-
Phosphocholine, glycerophosphocholine, and free choline are known as total choline compounds (tCho) [8, 116, 126], also known as choline phenotypes. Elevated levels of phosphocholine and total choline compounds have been observed in most cancers, including breast, ovarian, prostate, cervical, brain and endometrial cancers, which are characteristic of choline metabolites in cancer [127, 128, 129]. Currently, abnormal choline metabolism has been used as an endogenous biomarker for cancer [113]. Non-invasive imaging techniques such as positron emission tomography (PET) and high-resolution multinuclear magnetic resonance spectroscopy (MRS) can detect these changes in choline metabolism, and the choline phenotype is being used for tumor radio diagnosis, monitoring and prognosis, as well as the development of new therapies [9, 127, 130].
Positron emission tomography (PET) is a nuclear medicine modality that offers
excellent sensitivity and the potential to provide quantitative information on
biochemical processes within tissues or specific target macromolecules in
vivo. PET in combination with computed tomography (PET/CT) radiocholine (labeled
Currently, the evaluation of metabolic changes mainly relies on gas chromatography, liquid chromatography-mass spectrometry, and high-resolution multinuclear MRS technology [136]. Since the 1990s, MRS has made a great contribution to the characterization of phosphocholine metabolism in tumor cells and tissues, making it a potential indicator for tumor progression and therapeutic response [9, 137, 138]. Meanwhile, high resolution MRS can detect the homeostasis level and flux of metabolites in the biosynthesis and catabolism of phosphatidylcholine [139]. In the past 20 years, MRS has been widely used to study biochemical pathways in intact tissues and extracts from living tissues [8]. MRS can be used for noninvasive monitoring of various biologically important molecules in cells and tissues, highlighting important details critical to cell biochemistry. Therefore, MRS provides a noninvasive method to monitor the metabolites that play a central role in multiple pathways, measures the rate of reactions in these pathways in vivo, and studies how to control these rates in order to provide a given metabolic precursor and its required cofactors [8, 19, 139].
MRS uses different non-radioactive isotopes, such as
The advantage of MRS is that it does not need to separate and purify metabolites, thus avoiding the use of radioisotope substrates and fluorescence probe quenching [139]. Although it is limited by its low sensitivity, the application of ultra-high magnetic field intensity and the development of new technologies such as dynamic nuclear polarization (DNP) can reduce ameliorate this problem [144]. In addition, MRS has a unique advantage-high spectral resolution, and it can simultaneously detect and quantify the consumption of exogenously added substrates and the formation of products [137, 145]. Therefore, MRS can not only measure the basic level of intracellular metabolites in the samples, but also determine the activities of key enzymes involved in the metabolic pathways analyzed. In this way, MRS can simultaneously measure the activities of different enzymes in the same cell sample and quickly evaluate the rearrangement of metabolic flux under different experimental conditions [8].
In another study,
Liver cancer (including hepatocellular carcinoma and intrahepatic cholangiocarcinoma) is one of the most common malignant tumors in the world [149, 150]. In contrast to virally driven HCC, up to 50% of NAFLD-HCC occurs in patients without cirrhosis [50].
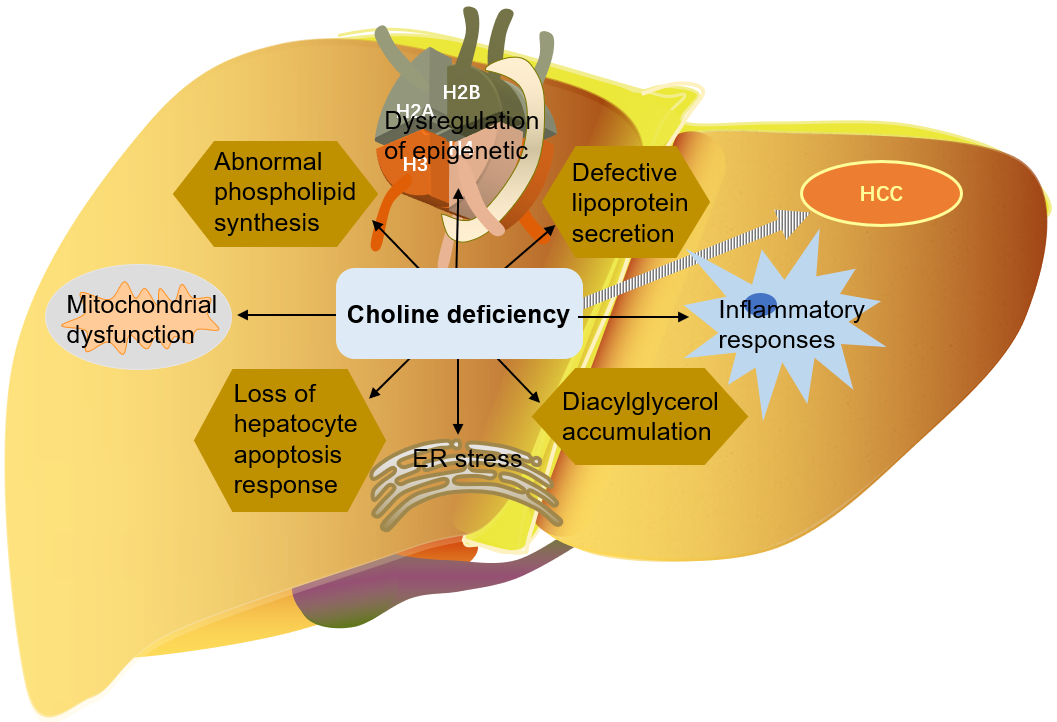
As mentioned above, choline is very important for liver health. Many mechanisms
by which choline deficiency exacerbates steatosis to liver cancer have been
found: abnormal phospholipid synthesis, defective lipoprotein secretion, DNA
oxidative damage and lipid peroxidation caused by mitochondrial dysfunction, ER
stress, loss of hepatocyte apoptosis response, and alteration of protein kinase C
signal transduction caused by diacylglycerol accumulation, etc. [9, 114, 116, 129]. Choline is an important part of mitochondrial membrane. Mitochondrial
dysfunction is the central mechanism of NAFLD-HCC. When choline levels are low,
mitochondrial bioenergy and fatty acid
The death of hepatocytes in the absence of choline can also cause inflammatory responses, accompanied by neutrophil/macrophage mediated production of reactive oxygen species and nitrogen [13, 153]. Studies have found that there is a link between choline metabolism and macrophage inflammation [154, 155]. Pro-inflammatory polarization increases choline uptake and phosphatidylcholine biosynthesis, thus promoting macrophages to respond appropriately to immune stimulation [155]. The increase of choline uptake provides the necessary amount of phosphatidylcholine for the synthesis of cell membrane (organelle membrane and plasma membrane), which is conducive to the packaging and secretion of cytokines [115]. In addition, regulation of phosphatidylcholine biosynthesis can reprogram the immune response in primary macrophages [24, 151].
As mentioned above, choline is involved in methyl metabolism and can be used as a donor of methyl. Since there are epigenetic mechanisms that rely on DNA or histome methylation to alter gene expression, choline also plays a role in epigenetic regulation. A choline-methyl deficient diet resulted in a decrease in S-adenosylmethionine concentration and an increase in S-adenosylhomocysteine concentration in the liver of male rats and mice [3]. A simultaneously low concentration of S-adenosylmethionine and high concentration of S-adenosylhomocysteine inhibit the activity of methyltransferase. As S-adenosylhomocysteine has a high affinity for DNA methyltransferases (DNMTs), it competes with S-adenosylmethionine to inhibit DNMTs enzyme activity. Thus, choline-methyl deficiency leads to decreased DNA methylation at many sites, including oncogene c-myc, but also increases methylation of other cytokines (including some tumor suppressor genes, such as p53). Many tumor suppressor genes are epigenetically regulated, including cell cycle regulation genes (p15 and p16), apoptosis genes (DAPK and APAF-1), cell adhesion genes (CDH1 and CDH3), and DNA repair genes (BRCA1 and hMLH) [45, 152]. Histone methylation was also affected by a diet deficient in methyl. Choline-methyl deficiency diet resulted in a decrease in histone H3-lysine 9-trimethylation (H3K9me3) and histone H4-lysine 20 trimethylation (H4K20me3) in the liver [156].
In addition, gut microbiota dysregulation is also involved in the development of
NAFLD-related liver cirrhosis and liver cancer. Dysregulation of gut microbiota
leads to a surge in inflammatory cytokines, such as TNF-
Compared with normal liver cells, hepatoma cells have drastically different epigenomes-hepatoma cells have gene specific DNA hypermethylation or hypomethylation, histone epigenetic markers changes, abnormal expression of DNA methyltransferase and histone modifying enzyme genes. Phosphatidylcholine is the precursor of lysophosphatidic acid (LPA), which regulates cell proliferation, differentiation, morphogenesis, and prevents apoptosis through G-protein-coupled transmembrane receptors. The genes encoding LPA signal receptors are regulated by epigenetic mechanisms. Abnormal methylation of LPA1 receptor gene was observed in rodents fed a choline-methionine deficient diet, with a pattern similar to that described in liver cancer [32].
Studies have shown that when the key gene in choline metabolism (betaine
homocysteine methyltransferase gene BHMT) is deleted in mice, these mice
will develop liver cancer [156]. It was also reported that the level of choline
kinase-
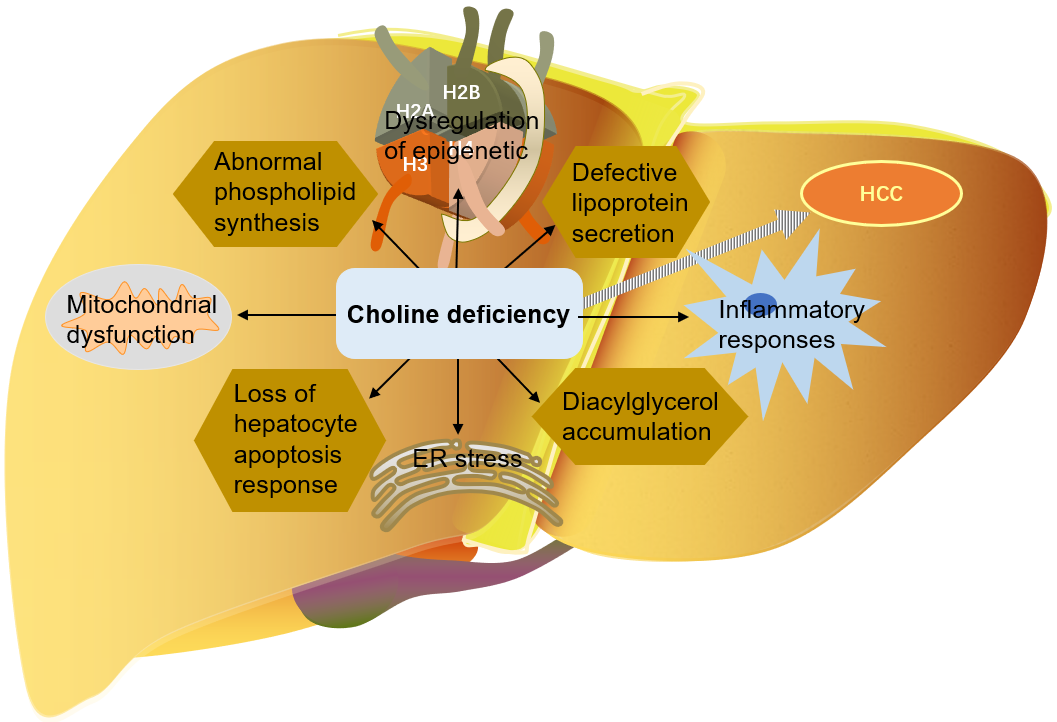 Fig. 6.
Fig. 6.The role of choline in the development of liver cancer. HCC, Hepatocellular carcinoma; ER, endoplasmic reticulum.
In addition to liver cancer, abnormal choline metabolism has been found in a variety of cancers [9]. Choline phosphate is involved in the malignant transformation of breast cancer, lung cancer, colon cancer, prostate cancer, neuroblastoma, liver lymphoma, meningioma, and other tumors through the RAS oncogene [22]. Many studies have shown that the expression and activity of lipases (including fatty acid synthase and choline kinase) are up-regulated in all stages of prostate cancer, and are associated with poor prognosis and survival rate. The cell membrane composition of normal cells and malignant cells is different-the relative content of choline phospholipid, which is mainly distributed in the outer lobe of cell membrane, decreased in all malignant tumors [12].
Choline kinase inhibitors can be used as a new type of antiproliferative and
anticancer drugs [158]. Several kinds of choline kinase inhibitors have been
developed: Hemicholinium-3 (HC-3) is a competitive inhibitor with similar
structure of Chk-
Another study found that the expression of choline kinase is partly dependent on the function of cyclooxygenase-2 (COX-2). COX-2 silencing can significantly reduce the levels of phosphocholine and tCho, significantly increase the lipid levels, and form lipid droplets in human breast cancer cells. Moreover, COX-2 silencing can transform the parental cell metabolite mode into a less invasive cancer cell metabolite mode. Therefore, COX-2 targeted therapy can prevent tumor invasion and metastasis [160].
Tumor development and metastasis involve epithelial mesenchymal transition (EMT). Some studies have shown that inhibition of phosphatidylcholine specific phospholipase C (PC-PLC) can lead to the loss of mesenchymal properties of metastatic breast cancer cells, indicating that PC-PLC plays an important role in the molecular pathway of maintaining mesenchymal like phenotype of metastatic breast cancer cells. Inactivation of PC-PLC may be a means to promote the differentiation of breast cancer cells and improve the effect of anti-tumor therapy [68].
Metabolites and enzymes in the choline metabolic cycle play a role in the
development of NAFLD, including increased uptake of fatty acids, reduced
mitochondrial fatty acid
The current treatment options for NAFLD encompass lifestyle changes (e.g., healthy diet and increased exercise), drug therapy, surgery, and liver transplantation [53, 161]. However, the currently available noninvasive diagnostic tests lack specificity and accuracy. Furthermore, liver biopsy, which has been considered the gold standard for assessing liver injury, remains invasive and carries inherent risks [15, 161, 162].
There is compelling evidence that NAFLD imposes a significant clinical and
economic burden, which is expected to further escalate [81, 163]. Ideally,
pharmacological interventions targeting NAFLD should impede its progression and
facilitate the reversal of liver injury. However, despite substantial investments
made by the pharmaceutical industry in recent years, there is currently no
approved treatment for NAFLD. Various drugs, such as FXR agonists, PPAR
NAFLD patients may develop advanced fibrosis and cirrhosis, or even liver cancer. Choline deficiency is involved in the pathogenesis of NAFLD-HCC, including abnormal phospholipid synthesis, defective lipoprotein secretion, mitochondrial dysfunction, ER stress, initiation of inflammatory reaction, loss of hepatocyte apoptosis, and changes of protein kinase C signal transduction caused by diacylglycerol accumulation. DNA oxidative damage and lipid peroxidation caused by mitochondrial dysfunction are the central mechanisms of NAFLD-HCC [13, 32]. Choline is additionally involved in epigenetic regulation [3, 32, 152], while dysregulation of the gut microbiota is implicated in NAFLD-related liver cirrhosis and liver cancer [28].
Besides liver cancer, choline metabolism is implicated in the development of
various other cancers, such as breast cancer, ovarian cancer, prostate cancer,
cervical cancer, brain cancer, and endometrial cancer [95, 96, 97]. Elevated
levels of phosphocholine and total choline (tCho) are distinctive choline
metabolites in cancer and are increasingly utilized as endogenous cancer
biomarkers [95, 96, 97]. Moreover, enzymes like Chk-
Choline kinase inhibitors, including HC-3, MN-58b, RSM-932A, and TCD-717, among others, are anticipated to emerge as novel anti-tumor therapeutics in the future. Nevertheless, there are currently no effective choline kinase inhibitors available for clinical use. Therefore, additional experimental and clinical validations are necessary to develop dependable choline kinase inhibitors [1, 126, 130].
In conclusion, choline metabolism plays a role in the pathogenesis of NAFLD and various cancers, including liver cancer, making it a viable target for future therapeutic interventions. At present, there are no effective therapeutic agents that specifically target choline metabolism for NAFLD and cancers in the clinical setting, necessitating further experimental and clinical validation.
Not applicable.
Conceptualization, XC, WYQ, CX and RZC; methodology, XC, WYQ, XQM, LLR; writing—original draft preparation, XC, WYQ, CX and RZC; collecting literature, draw charts—MQF, SH; writing—review and editing, XC, MQF, SH, CX and RZC. All authors contributed to editorial changes in the manuscript. All authors have read and agreed to the published version of the manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.