







1 Department of Radiation Oncology, Ningbo Medical Center Lihuili Hospital, 315000,Ningbo, Zhejiang, China
2 Department of Urology, Ningbo Urology and Nephrology Hospital, 315100 Ningbo, Zhejiang, China
Abstract
Renal cell carcinoma (RCC) is a prevalent and aggressive kidney cancer with notable metastatic potential. While radiotherapy is effective for treating metastatic RCC, the emergence of radioresistance presents a major challenge. This study explores the role of DLX5, previously identified as an oncogene in various cancers, in the development of radioresistance in RCC.
Distal-less homeobox 5 (DLX5) expression was measured using western blot analysis. To study the effects of DLX5, its expression was knocked down in 786-O and Caki-1 RCC cell lines through si-DLX5 transfection, and the impact of DLX5 on RCC cell proliferation and radioresistance was assessed using cell counting kit-8 (CCK-8), 5-Ethynyl-2′-deoxyuridine (EdU) incorporation assay, flow cytometry, colony formation, immunofluorescence, and western blot assays. The underlying mechanisms were explored through western blot, colony formation, and CCK-8 assays. In vivo effects were examined using a xenograft mouse model.
In silico results showed increased DLX5 levels in RCC tissues. Similarly, DLX5 expression was elevated in RCC cell lines. Silencing DLX5 reduced RCC cell proliferation and induced apoptosis in vitro. Additionally, DLX5 knockdown decreased radioresistance and increased DNA damage in RCC cells. Mechanistic studies revealed that DLX5 promotes radioresistance through the upregulation of c-Myc. In vivo, DLX5 silencing impeded tumor growth and reduced radioresistance.
DLX5 contributes to RCC cell growth and radioresistance by upregulating c-Myc expression, highlighting its potential as a target for overcoming radioresistance in RCC.
Graphical Abstract
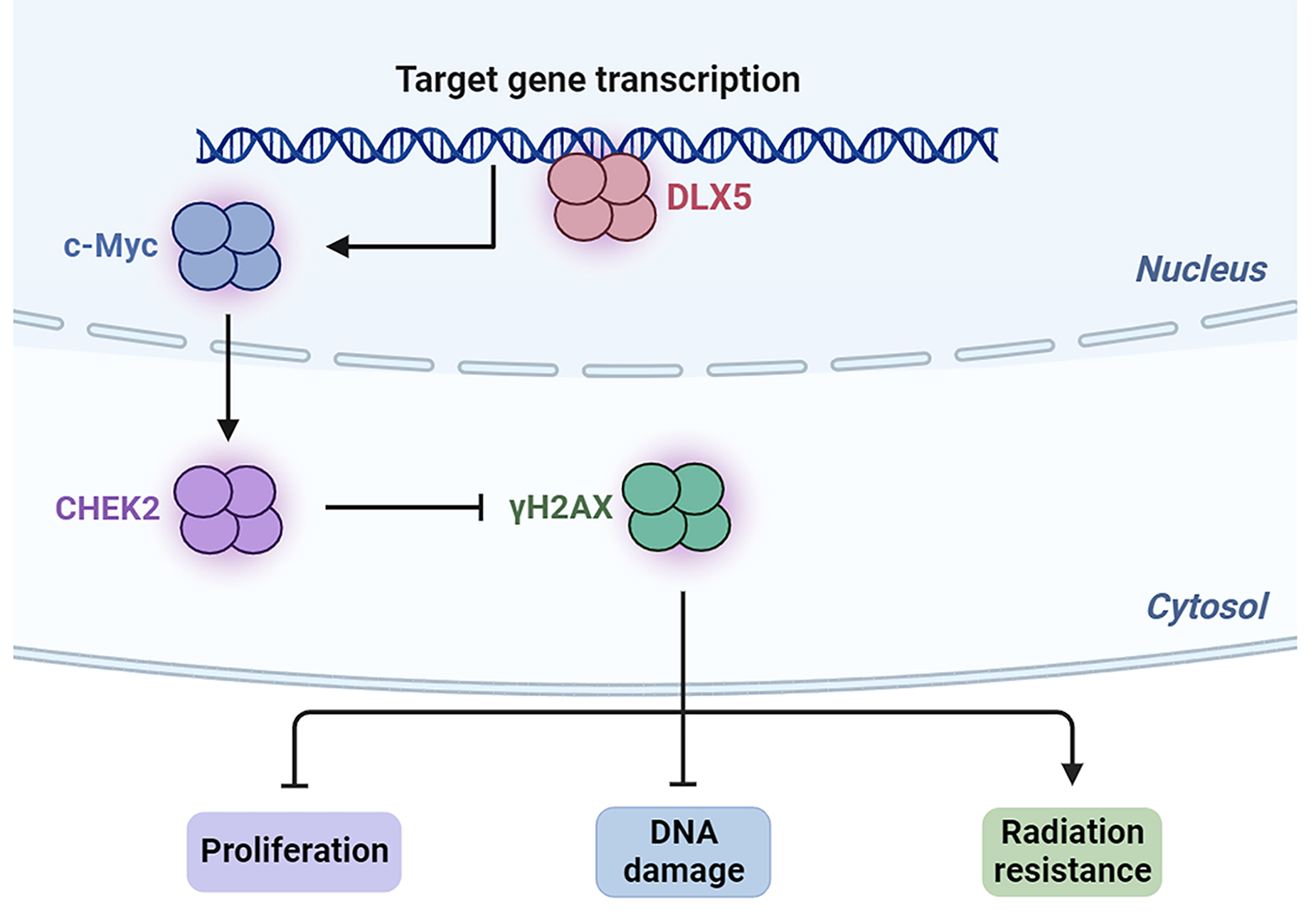
Keywords
- renal cell carcinoma
- DLX5
- radioresistance
- c-Myc
Renal cell carcinoma (RCC) represents the most prevalent type of renal tumor, comprising nearly 85% of all kidney tumors and 3% of all adult malignancies, and its incidence and mortality rates have been rising in recent years [1, 2]. Current clinical treatment options, including surgery, targeted therapy, and immunotherapy, have improved the 5-year overall survival rate to 70%–80% [3, 4]. However, 30% of patients eventually develop metastatic RCC following surgical intervention [5]. Therefore, elucidating the underlying mechanisms of RCC and identifying potential therapeutic targets is essential.
Radiotherapy is designed to damage the genome of tumor cells, leading to cell death through the generation of irreparable DNA double-strand breaks (DSBs) [6]. Enhanced repair of DNA DSBs can contribute to resistance to ionizing radiation (IR) [7]. c-Myc plays a significant role in various biological processes in kidney cancer, including the regulation of metabolism [8]. c-Myc has been shown to interact with and regulate the promoters of genes involved in DSB repair in prostate cell lines [9]. In this regard, Wang et al. [10] demonstrated that c-Myc binds to the promoters of CHK1 and CHK2, essential regulators of the DNA damage response (DDR), thereby enhancing radioresistance in stem cell-like populations of nasopharyngeal carcinoma cells. Despite these findings, little is known about the effects of c-Myc in RCC radioresistance.
The distal-less homeobox (DLX) family (DLXs), which includes DLX1 through DLX6, is related to the Drosophila distal-less gene and is represented by three clusters: DLX1/DLX2, DLX3/DLX4 and DLX5/DLX6 [11]. DLXs are involved in cell differentiation and proliferation, primarily during embryonic development, including in the nervous system, appendages, hematopoiesis, and branchial arches [11, 12]. In addition, there is increasing evidence indicating that DLXs are deregulated in various tumors, suggesting their potential as therapeutic targets [13, 14]. DLX5, in particular, has demonstrated potent oncogenic activity in oral squamous cell carcinoma (OSCC) [15], ovarian cancer [16], and lung cancer [17]. However, the role of DLX5 in RCC is not yet understood. Notably, DLX5 has been shown to directly bind the c-Myc promoter in lung tumor cells [17]. This raises the hypothesis that DLX5 may influence radioresistance in RCC through interaction with c-Myc.
Therefore, this study aims to investigate the role of DLX5 in RCC development and radioresistance both in vitro and in vivo. The results may provide a theoretical foundation for advancing RCC treatment strategies.
DLX5 expression levels in kidney renal clear cell carcinoma (KIRC) and normal tissues were evaluated using the UALCAN (http://ualcan.path.uab.edu/index.html), TIMER (https://cistrome.shinyapps.io/timer/), and GEPIA (http://gepia2.cancer-pku.cn) databases.
RCC cell lines 786-O (CL-0010), A-498 (CL-0254) and Caki-1 (CL-0052), and HK-2 (CL-0109) cells were purchased from Procell (Wuhan, China). A-498 and HK-2 cells were cultured in Minimum Essential Medium (MEM, PM150410, Procell), while 786-O and Caki-1 cells were grown in RPMI-1640 (PM150110, Procell) and McCoy’s 5A media (PM150710, Procell), respectively, with 10% fetal bovine serum (FBS, 164210-50, Procell) and 1% penicillin/streptomycin (PB180120, Procell). Cells were authenticated by short tandem repeat (STR) profiling, tested negative for mycoplasma, and maintained in a 37 °C, 5% CO2 incubator.
Two small interfering RNAs (siRNAs) targeting DLX5 (si-DLX5#1 and si-DLX5#2) and corresponding negative controls (si-NC) were synthesized by GenePharma (Shanghai, China). The sequences of si-DLX5#1, si-DLX5#2, and si-NC were 5′-AGCUAUAGUCGGCAUAAGCUU-3′, 5′-GUGCAGCCAGCUCAAUCAA-3′, and 5′-UUCUCCGAACGUGUCACGU-3′, respectively. Overexpression of c-Myc was achieved by transfecting pcDNA vector plasmids containing c-Myc (NM_005221.5) sequences (OE-c-Myc). The following transfections were performed: si-DLX5#1, si-DLX5#2, si-NC, si-NC+empty pcDNA vector plasmids (vector), si-DLX5#1+vector, and si-DLX5#1+OE-c-Myc into 786-O and Caki-1 cell lines using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA), and the cells were then collected for subsequent analyses 48 hours later.
Total proteins were extracted using RIPA buffer (R0010,
Solarbio, Beijing, China) and quantified with a BCA protein assay (ab102536,
Abcam, Cambridge, UK). Proteins (20 µg) were separated by SDS-PAGE
and transferred to PVDF membranes (EMD Millipore, Burlington, MA, USA). Membranes were blocked with
5% skim milk (D8340, Solarbio) for 1 hour at room temperature, then incubated
overnight at 4 °C with primary antibodies against DLX5 (1:1000,
ab64827), gamma histone H2AX (
Cells (3000 per well) were plated in 96-well plates and cultured overnight at 37 °C with 5% CO₂. After treatments, 10 µL of CCK-8 reagent (C0037, Beyotime) was added to each well, the plates were incubated for 2 hours at 37 °C, and optical density (450 nm) was measured using a microplate reader (Thermo Fisher Scientific, Waltham, MA, USA).
Cells (6
Cells were stained with propidium iodide (PI) and fluoresceinisothiocyanate (FITC)-Annexin V (C1062S, Beyotime), and then washed with cold PBS (C0221A, Beyotime). Apoptosis was assessed using a FACScan flow cytometer (BD Biosciences, NJ, USA), and data was analyzed using BD CellQuest Pro software (version 5.1, BD Biosciences).
Cells were irradiated in ambient air with total doses of 0, 2, 4, 6, and 8 Gray (Gy) using a JL Shepherd Mark 1–68 137Cs irradiator. Mice with subcutaneous tumors were irradiated with 10 Gy using an XRad 320 irradiator (Precision X-Ray, 250 kV/s, 15 mA) following anesthesia with isoflurane (R510-22, RWD LIFE SCIENCE, Shenzhen, China).
Cells (800 per well) were plated in 6-well plates and incubated at 37 °C for 14 days. After fixation with 4% paraformaldehyde (P0099, Beyotime), cells were stained with 0.1% crystal violet (C0121, Beyotime) for 30 minutes. Colonies with 50 or more cells were counted manually and photographed. Plating efficiency (PE) was calculated by dividing the number of colonies in the control group by the number of cells plated. The survival fraction was determined by dividing the number of colonies by the product of the number of cells plated and the PE.
Cells were plated on glass coverslips and cultured overnight. After irradiation
with 4 Gy, cells were collected at 0, 1, 4, and 8 hours. For immunofluorescence,
cells were fixed with 4% paraformaldehyde (P0099, Beyotime) for 15 minutes,
washed with PBS (C0221A, Beyotime), and permeabilized with 0.5%
Triton X-100 (P0096, Beyotime) for 20 minutes. Following
overnight incubation with anti-
Four-week-old BALB/c
nude mice (Vital River, Beijing, China) were housed in a temperature-controlled
specific pathogen free (SPF) facility with a 12-hour light-dark cycle. Mice were
randomly assigned to four groups (n = 6 each): sh-NC, sh-DLX5#1, 10 Gy, and
sh-DLX5#1+10 Gy. Each group had six mice throughout the study. The left flank of
mice in the sh-NC and sh-DLX5#1 groups were subcutaneously injected with 2
Tumor samples were fixed in 4% formaldehyde (P0099, Beyotime), dehydrated, and embedded in paraffin (YA0012, Solarbio). Antigen retrieval was done with 10 mM sodium citrate buffer (pH 6.0, P0083, Beyotime) at 94 °C for 15 minutes. Sections were blocked with 1% bovine serum albumin (BSA) (ST023, Beyotime), then incubated with primary antibodies against DLX5 (1:100, ab64827, Abcam), c-Myc (1:100, ab32072, Abcam), and Ki-67 (1:100, ab15580, Abcam). Biotinylated secondary antibodies (1:2000, ab64256, Abcam) were used, and sections were counterstained with hematoxylin (C0105S, Beyotime). The samples were imaged with a light microscope (Olympus).
Data are presented as mean
Pan-cancer analysis revealed a significant increase in DLX5 expression across various cancer types, including KIRC (Fig. 1A). Specifically, DLX5 was markedly upregulated in KIRC samples compared to normal kidney tissue samples (Fig. 1B,C). Additionally, high levels of DLX5 were validated in RCC cell lines (Fig. 1D). Notably, DLX5 expression was higher in 786-O and Caki-1 cells compared to A-498 cells, leading to the selection of 786-O and Caki-1 for further experiments. These findings indicate that DLX5 is highly expressed in RCC.
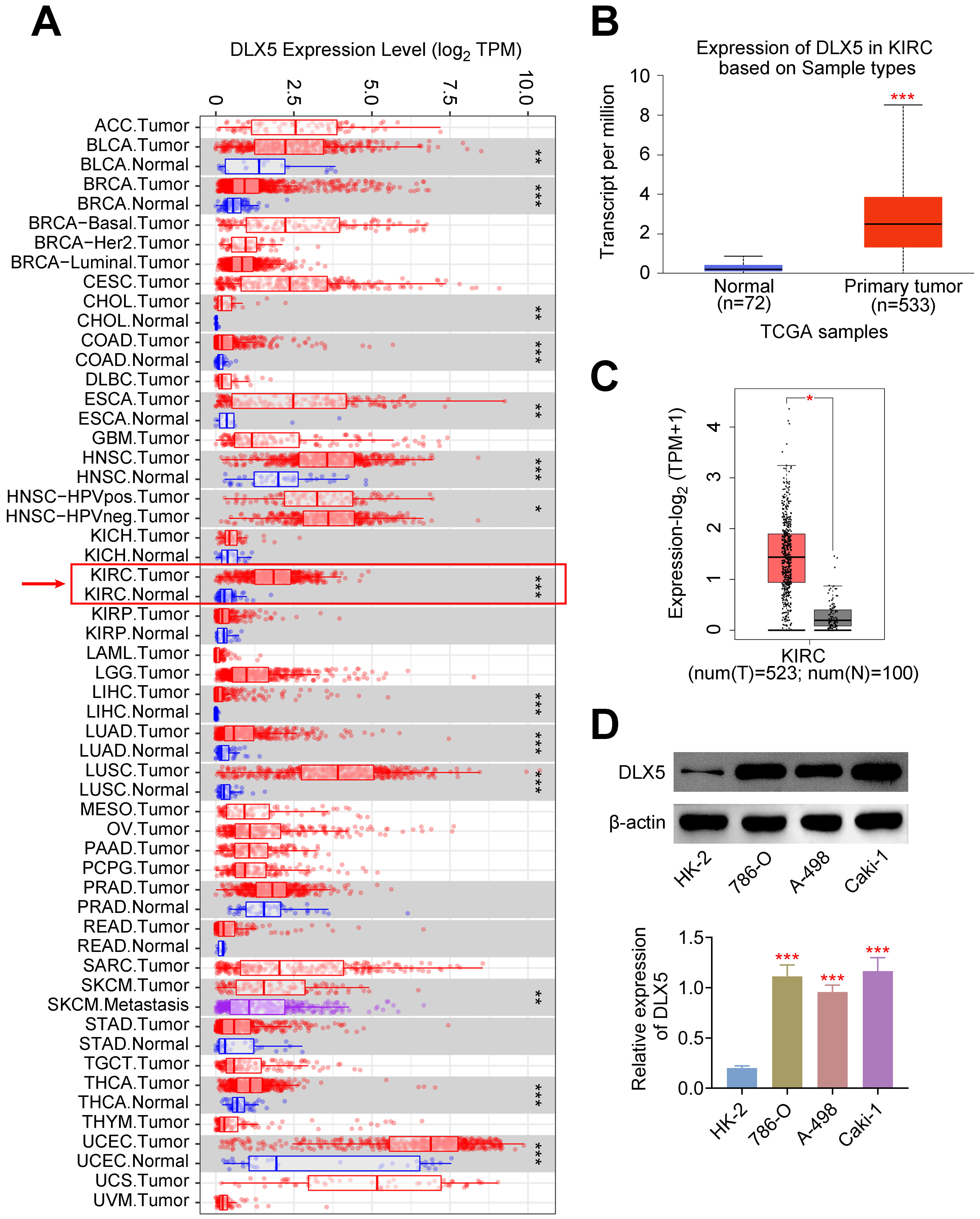 Fig. 1.
Fig. 1.
Elevated expression of DLX5 in renal cell carcinoma
(RCC). (A) Pan-cancer analysis demonstrated increased DLX5 expression in kidney
renal clear cell carcinoma (KIRC). *p
Given the high DLX5 expression in RCC cells, we used two siRNAs targeting DLX5 (si-DLX5#1 and si-DLX5#2) to lower DLX5 levels in 786-O and Caki-1 cells (Fig. 2A). DLX5 silencing significantly reduced cell proliferation, as shown by CCK-8 (Fig. 2B) and EdU assays (Fig. 2C and Supplementary Fig. 1), and increased apoptosis, as indicated by flow cytometry (Fig. 2D). Additionally, DLX5 knockdown enhanced p53 protein expression (Fig. 2E), suggesting DLX5’s role in regulating p53-mediated apoptosis and RCC cell proliferation. Thus, DLX5 downregulation impairs RCC cell growth and promotes apoptosis.
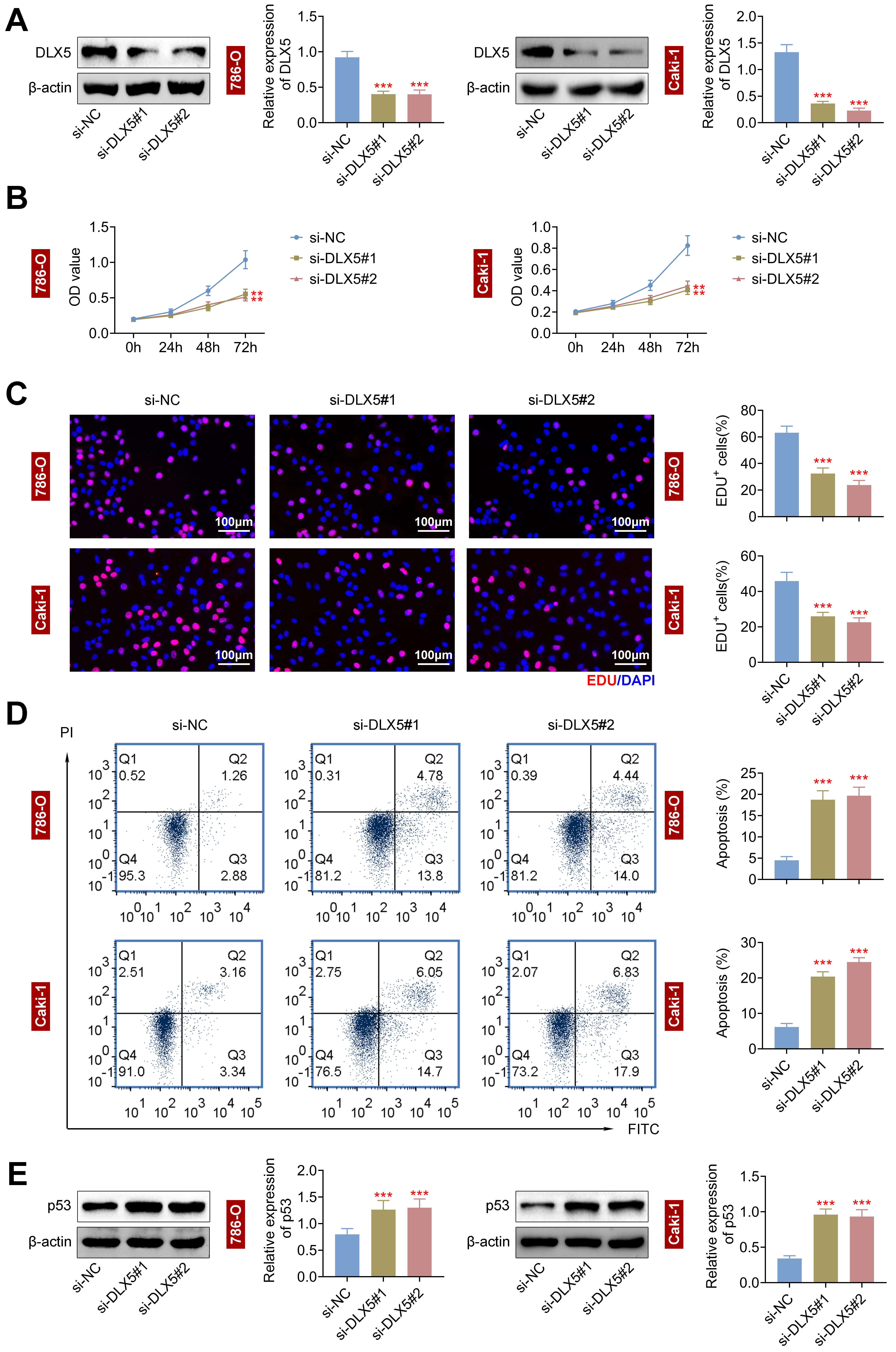 Fig. 2.
Fig. 2.
Silencing of distal-less homeobox 5 (DLX5) represses cell proliferation and enhances
apoptosis in RCC cells. (A) DLX5 levels were decreased in 786-O and Caki-1 cells
using si-DLX5#1 and si-DLX5#2, with
The impact of DLX5 on radioresistance was assessed in 786-O and Caki-1 cells.
Following transfection with si-DLX5#1 and si-DLX5#2, both cell lines exhibited
a dose-dependent reduction in survival rates (Fig. 3A). Notably, the survival
rates of 786-O and Caki-1 cells were significantly diminished following 8 Gy of
radiation (RI). Additionally, cell viability was markedly reduced at a total dose
of 4 Gy (p
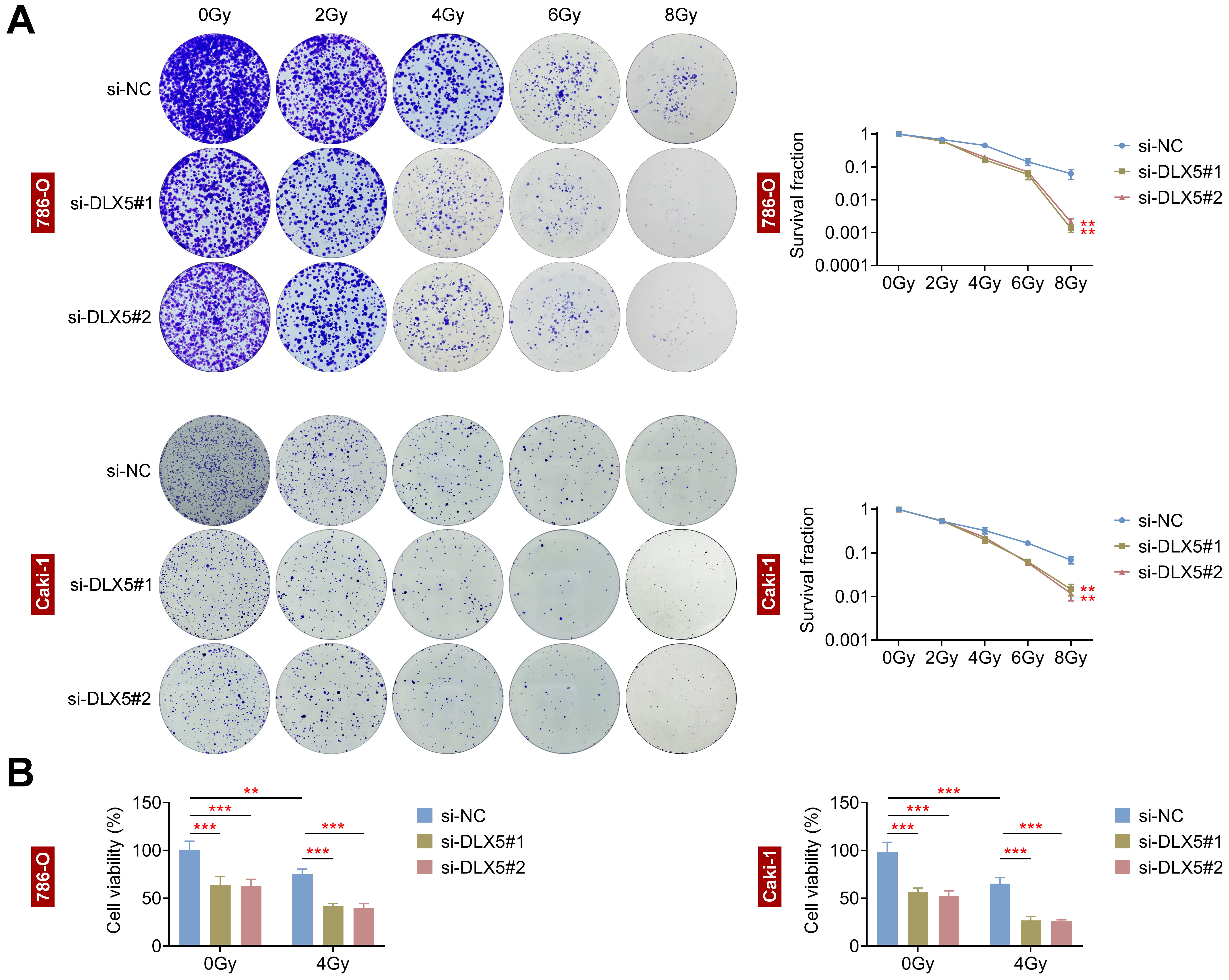 Fig. 3.
Fig. 3.
Interference with DLX5 inhibits radioresistance in RCC cells.
(A) Colony formation assays assessed survival fractions of 786-O and Caki-1 cells
transfected with si-NC, si-DLX5#1, and si-DLX5#2 following irradiation at 0, 2,
4, 6, and 8 Gray (Gy). **p
To examine DLX5’s impact on DNA damage, we analyzed 786-O and Caki-1 cells. DLX5
downregulation significantly increased
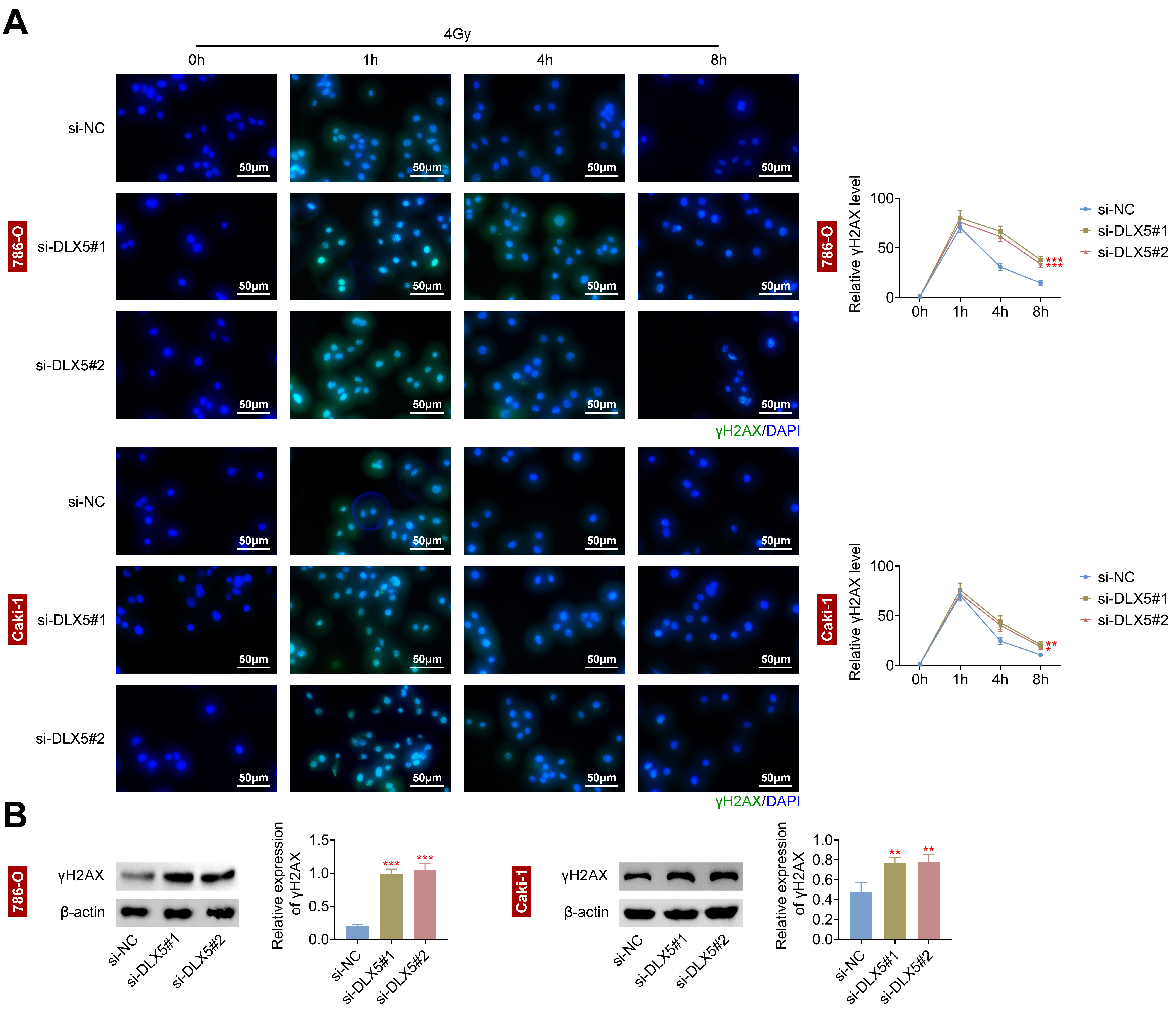 Fig. 4.
Fig. 4.
Interference with DLX5 increases DNA damage in RCC cells. (A)
The relative gamma histone H2AX (
c-Myc is known to interact with and modulate the promoters of DNA double-strand
break (DSB) repair genes, thereby enhancing radioresistance in tumor cells [9, 10]. Additionally, DLX5 has been shown to directly bind to the c-Myc promoter in
lung tumor cells [17], suggesting that DLX5 may influence radioresistance via the
regulation of c-Myc. In this study, we found that DLX5 modulated c-Myc
expression, as silencing DLX5 significantly reduced the 4 Gy-induced relative
protein expression of c-Myc in 786-O cells (Fig. 5A). Further validation was
performed by co-transfecting 786-O cells with an overexpression plasmid of c-Myc
and si-DLX5#1. Results showed that overexpression of c-Myc significantly rescued
the reduced c-Myc expression induced by si-DLX5#1, both with and without
radiation treatment (Fig. 5B). Moreover, c-Myc overexpression in 786-O cells
restored the decreased survival rate (Fig. 5C) and cell viability (Fig. 5D)
caused by DLX5 silencing. Additionally, increased c-Myc expression markedly
reduced the
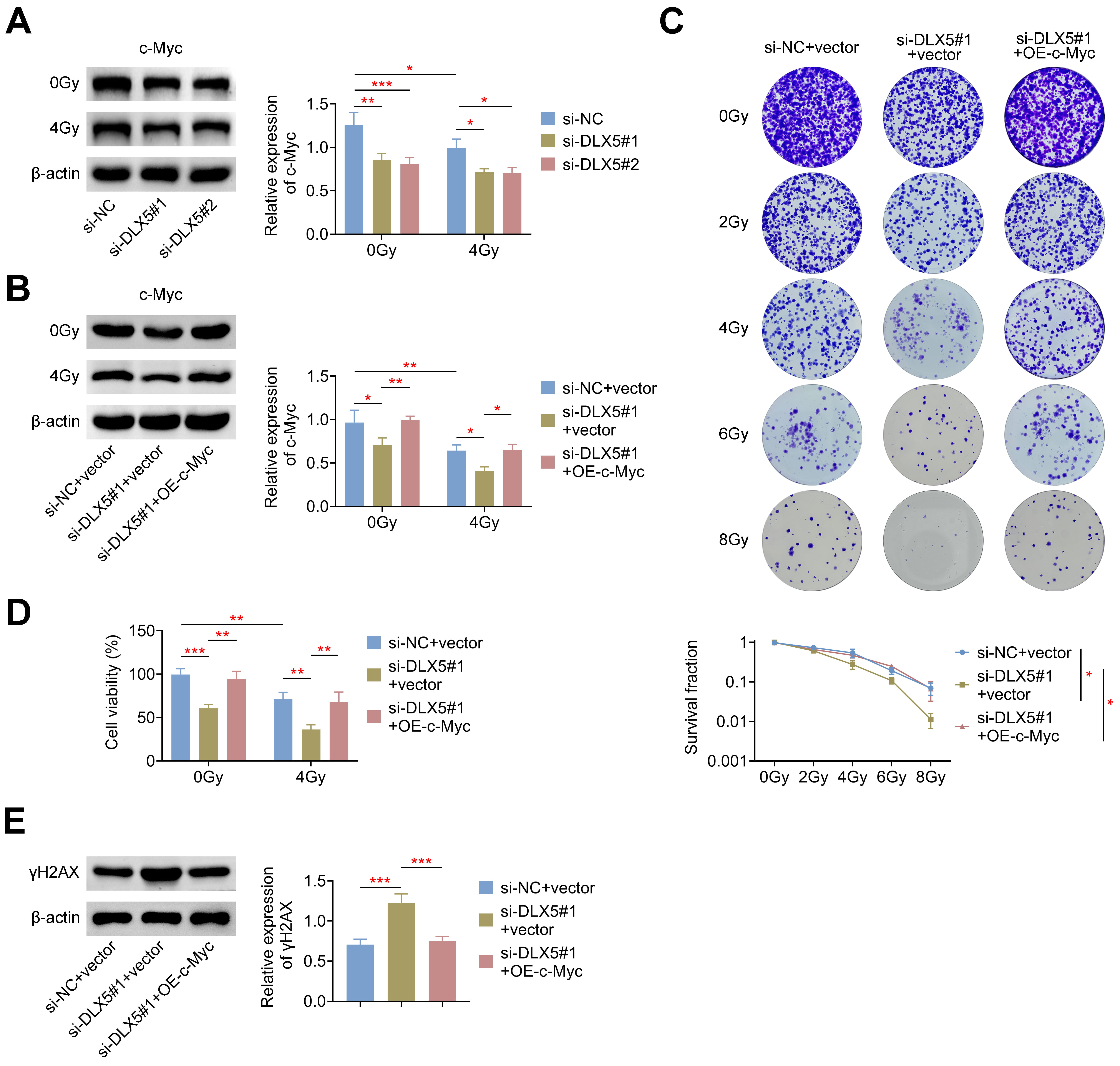 Fig. 5.
Fig. 5.
DLX5 enhances radioresistance through c-Myc. (A) c-Myc protein
levels were measured in 786-O cells transfected with si-NC, si-DLX5#1, or
si-DLX5#2, normalized to
Lastly, we evaluated the role of DLX5 in tumor progression and radioresistance
in nude mice. DLX5 knockdown or irradiation with 10 Gy significantly reduced
tumor volume and weight. This reduction was further pronounced when DLX5
silencing was combined with 10 Gy irradiation (Fig. 6A). Similar trends were
observed in the expression levels of ki-67 and c-Myc, which were significantly
decreased under these conditions (Fig. 6B). Additionally, both DLX5 silencing and
10 Gy irradiation led to a marked increase in
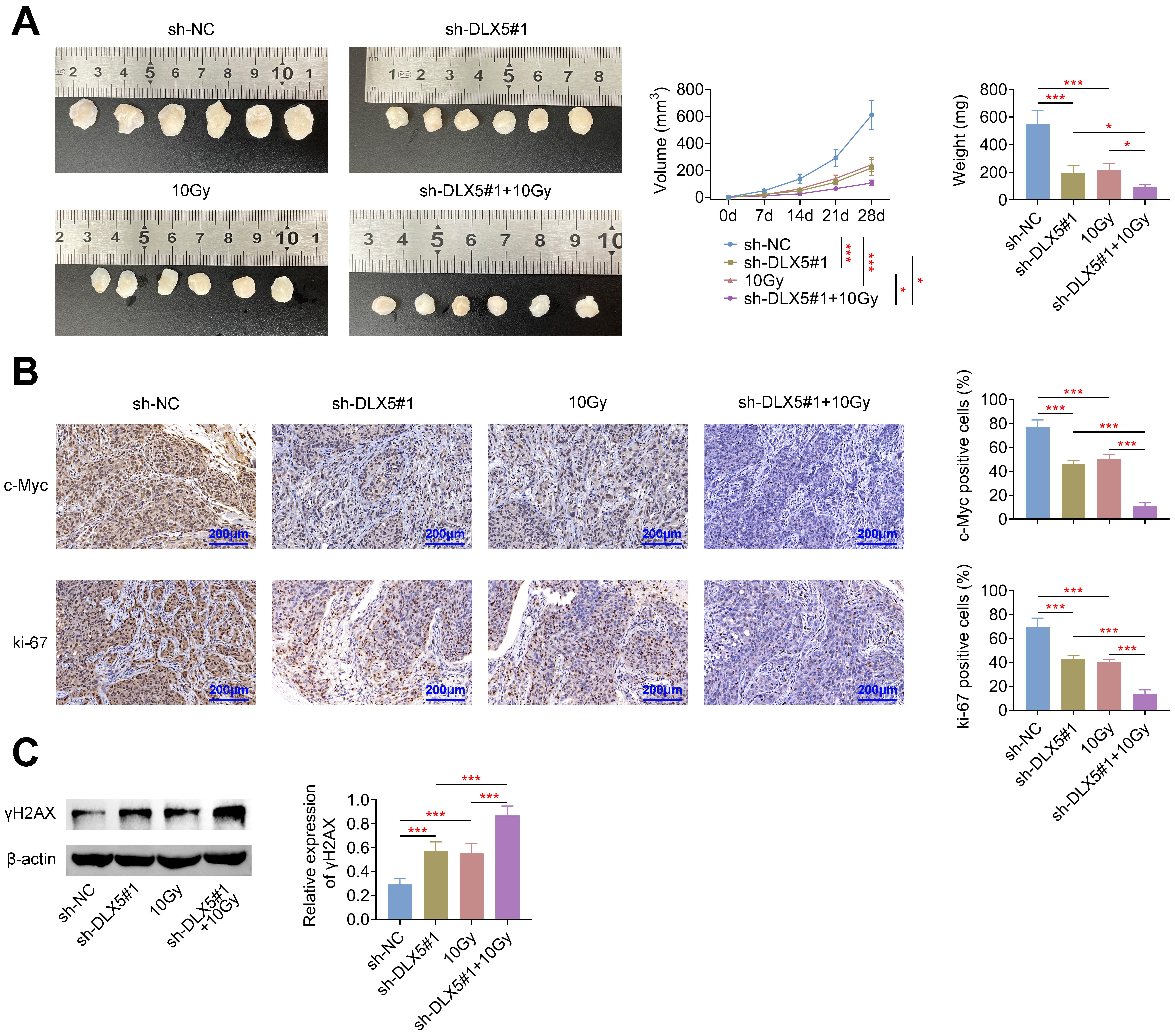 Fig. 6.
Fig. 6.
Silencing of DLX5 inhibits radioresistance in vivo.
Nude mice were inoculated subcutaneously with 2
RCC is a malignant kidney tumor characterized by a rising incidence and mortality rate. The prognosis of RCC is further compromised by metastasis, which exacerbates the disease outcome [5]. Despite the potential of radiotherapy for managing metastatic RCC, radioresistance has emerged as a significant clinical challenge [18]. DLX5, an oncogene implicated in various cancers, has been shown to play a critical role in tumorigenesis [15, 16, 17]. Our study demonstrates that DLX5 expression is significantly elevated in RCC, and its downregulation inhibits cell proliferation while promoting apoptosis. Additionally, DLX5 silencing reduces radioresistance and enhances DNA damage in RCC cells. Mechanistically, DLX5 appears to facilitate radioresistance through the upregulation of c-Myc. Moreover, the suppression of DLX5 impedes tumor growth and radioresistance in vivo. Collectively, DLX5 can lead to RCC progression and radioresistance by modulating c-Myc expression.
DLX5 expression is elevated in various tumors, including OSCC [15], ovarian cancer [16] and lung cancer [17]. Elevated DLX5 levels are generally associated with adverse clinical outcomes. For instance, high DLX5 expression was found to be correlated with increased tumor growth both in vitro and in vivo, advanced TNM stages, lymph node metastasis, poor cellular differentiation, and worse prognosis [15]. In ovarian cancer, DLX5 upregulation has been linked to enhanced cell proliferation and tumor size [16]. Consistent with these observations, our study shows that high DLX5 expression in RCC cells promotes growth and radioresistance and that DLX5 downregulation increases apoptosis and reduces tumor growth in vitro and in vivo, highlighting its role in RCC progression.
In addition, our study reveals that DLX5 interference impedes radioresistance in
RCC cells. The primary mechanism of radiation damage in radiotherapy involves DNA
damage, which disrupts the cell cycle and activates various signaling pathways to
repair the damaged DNA [19]. The effectiveness of radiation therapy in treating
malignancies can be enhanced by modulating DNA damage repair processes, which are
influenced by the interplay between DNA damage response (DDR) activation and
repair mechanisms [20, 21]. A key feature of DDR activation is the accumulation
of
This study acknowledges several limitations. Firstly, the clinical parameters associated with DLX5 will be explored in future research to further validate the clinical relevance of our findings. Additionally, while our in vitro results are promising, further validation through in vivo experiments involving the overexpression of c-Myc is necessary to confirm the role of DLX5 in RCC radioresistance.
In summary, the findings demonstrate that DLX5 expression is significantly increased in RCC. Loss-of-function assays revealed that DLX5 enhances RCC cell growth and radioresistance, with mechanistic insights indicating that DLX5 promotes these effects through the upregulation of c-Myc. These results support DLX5 as an essential player for RCC progression and radioresistance, identifying it as a potential marker for diagnosis and therapeutic targeting. Furthermore, targeting DLX5 or its downstream pathways may offer new strategies for overcoming radioresistance in RCC.
All data containing in the study are available upon requirement by contacting the corresponding author on reasonable request.
DH, ML and XC contributed to the study conception and design. Material preparation and the experiments were performed by DH. Data collection and analysis were performed by ML. The first draft of the manuscript was written by XC and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
All animal experiments were approved by the Ethics Committee of Zhejiang Huitong Test & Evaluation Technology Group Co., Ltd (Approval No. HT-2023-LWFB-0018) for the use of animals and conducted in accordance with the National Institutes of Health Laboratory Animal Care and Use Guidelines. The animal experiment complies with the ARRIVE guidelines and in accordance with the National Institutes of Health guide for the care and use of Laboratory animals (NIH Publications No. 8023, revised 1978).
Not applicable.
This study was supported by the Medical Science and Technology Project of Zhejiang Provincial Health Commission (Grant No.2024KY1605).
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/j.fbl2911400.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.