1. Introduction
Inflammatory response is a key mechanism driving cardiovascular disease, and a
critical risk factor for the development of cardiovascular conditions. In
response to heart injury, necrotic cardiac cells release a variety of
damage-associated molecular patterns (DAMPs) that activate NLR family pyrin
domain containing 3 (NLRP3) inflammasomes in cells of the innate immune system
[1]. The NLRP3 inflammasome is a key multiprotein signaling platform for
activation of inflammatory responses, and plays an important role in heart injury
by activating caspase-1 for the subsequent maturation of pro-inflammatory
cytokines, interleukin-1 (IL-1) and interleukin-18 (IL-18) [2].
Both IL-1 and IL-18 are members of the pro-inflammatory cytokine IL-1
superfamily and are often found in the serum during inflammatory stimulation.
Cardiac-derived IL-1 can impair cardiac contractility by inducing a
calcium leak from the sarcoplasmic reticulum, ultimately promoting cell death and
tissue remodeling [3].
Arginine vasopressin (AVP) is a vasopressive peptide composed of nine amino
acids in mammals. As a neurohormone and hemodynamics factor AVP maintains fluid
balance homeostasis, vascular tonus, and the regulation of the endocrine stress
system [4]. These physiological effects are mediated in humans via binding to at
least three G-protein-coupled receptors (GPCRs) subtypes, which have been
identified as VR, VR and VR (also termed V3R) [5]. However,
only VR is found in cardiac myocytes and cardiac fibroblasts [6]. AVP can
be synthesized in the heart and elicits local and potentially systemic effects
via cardiac paracrine [7] signaling. Of note, AVP levels are elevated relative to
the severity of heart failure or left ventricular dysfunction [8]. In our earlier
studies, AVP induced the expression of IL-6 in rat cardiac fibroblasts [9]. Both
IL-6 and IL-1 belong to the interleukin family, so it is possible that
AVP could induce the expression of IL-1 in heart cells and whether this
process was related to the NLRP3 inflammasome.
-arrestins are commonly expressed cytoplasmic adapter proteins, which
function, along with heterotrimeric G proteins, as critical regulators and signal
transducers for GPCRs [10]. -arrestins mediate neuropeptide-driven
inflammatory disease and GPCR-mediated inflammation [10, 11]. Specifically,
-arrestins regulate toll-like receptor-interleukin 1 receptor
(TLR-IL-1R)-induced signal transduction in macrophages which promotes
inflammation [12]. Moreover, TLRs upregulate the NLRP3 inflammasome by inducing
the activation of NF-B. In addition, -arrestin1, which is 78%
identical to -arrestin2 at the amino acid level, is essential for the
full activation of NLRP3 [13]. Earlier studies from our group have shown that
-arrestin2 regulates inflammation by activating the AVP-mediated
ERK-NF-B (ERK: extracellular regulated protein kinases) signaling pathway which induces expression of IL-6
[6]. In this study, the role of AVP in regulating the expression of
IL-1, as well as the roles of -arrestin2 and the NLRP3
inflammasome in regulating this process, were further explored both in
vivo and in vitro and this work may provide a new therapeutic approach
for the treatment of cardiovascular diseases.
2. Materials and Methods
2.1 Animals
The adult Sprague-Dawley rats were obtained from the Animal Center of Nantong
University (Nantong, China). The -arrestin2 knockout mice were obtained
from the laboratory of Dr. Lefkowitz (Duke University, Durham, USA). Male rats
and mice (8–12 weeks old) were administered with 2 g/g of AVP via
tail vein injection. Heart tissues were harvested and RNA and protein obtained at
designated timepoints after AVP administration. The study was approved by the
Board of Nantong University Animal Care and Use.
2.2 Materials
NF-B p65 antibody (D14E12), p-NF-B antibody (93H1),
anti-rabbit IgG, anti-mouse IgG, GAPDH (glyceraldehyde-3-phosphate dehydrogenase) antibody, and -arrestin1/2
antibody (D24H9) were purchased from Beijing Green Herbs Biological Co. LTD
(Beijing, China). NLRP3 antibody (ab214185) and IL-1 antibody (ab205924)
were purchased from Abcam (Cambridge, UK). The primers for murine IL-1
and GAPDH were purchased from Shanghai Sangon Biotech (Shanghai, China). Total RNA
Extraction Kit and Total DNA Extraction Kit were from Zymo Research (Beijing,
China). Rat IL-1 ELISA Kit (E-EL-R0017c) and mouse IL-1 ELISA
Kit (E-EL-M0079c) were obtained from Elabscience (Wuhan, China). PDTC (S1808) was
obtained from Shanghai Beyotime Biotechnology (Shanghai, China). ShRNA that target
-arrestin2 (shRNA898, shRNA899, shRNA900) was purchased from Shanghai
Genechem Co.Ltd (Shanghai, China).
2.3 Methods
2.3.1 Isolation and Culture of Cardiomyocyte and Cardiac
Fibroblasts
Adult rat cardiac fibroblasts (ARCFs) were obtained from the hearts of adult
(250–300 g) male Sprague Dawley rats after anesthesia with 10% chloralhydrate
(0.5 mL/kg body weight) as previously described [9]. Experiments were
consistently performed on passage 3–8 cells. Adult rat cardiac myocytes (ARCMs)
were isolated and cultured in serum-free Medium199 with 100 U/mL
penicillin-streptomycin solution for 2–4 h before passaging as previously
described [14].
2.3.2 Lentiviral Infection
ARCFs were washed twice with sterile PBS (phosphate buffer solution) and infected with lentivirus encoding
shRNA targeting -arrestin2 (shRNA898, shRNA899, shRNA900) at a MOI = 50.
Cells were subsequently incubated in fresh medium for 12 h, and the transfection
efficiency was observed under a fluorescence microscope after 72 h.
2.3.3 Propidium Iodide Assay
A propidium iodide (PI) assay was used to quantify cardiomyocyte death (ST512,
Shanghai, China). Cardiomyocytes were rinsed with PBS after AVP treatment and
incubated with Hoechst 33258 (C1018, Beyotime, China) for 15–30 min at 37 °C
for cell counting. The cells were then rinsed three times with PBS and incubated
with PI for 20–30 min at 37 °C to count dead cells. A Leica fluorescence
microscope was used to capture images.
2.3.5 Immunofluorescence
Mouse hearts were fixed in 4% paraformaldehyde and embedded at 4 °C overnight.
The hearts were cut into 5m of tissue slices with a tissue slicer and were
fixed in the cover slips. After wax removal and rehydration of tissue slices, the
myocardium samples were incubated with primary antibody to IL-1 or NLRP3
diluted in 0.4% Triton X-100 for permeabilization. Following incubation with
primary antibody, slides were washed 3 times with permeabilizing buffer, and the
slides were then labeled with RITC (rhodamine isothiocyanate)—labeled goat
anti-mouse IgG secondary antibody. Fluorescence density was assayed using Image J
software version 1.8.0.112 (NIH, Bethesda, MD, USA).
2.3.6 Quantitative PCR (qPCR) for Analyzing IL-1 mRNA
Levels
The level of the IL-1 and GAPDH RNA in ARCMs was quantified by qPCR.
Total RNA from cultured cardiomyocytes was extracted using Trizol reagent
(Promega, Madison, WI, USA) according to the manufacturer’s protocol. The
Step-one Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) was used
to amplify the cDNA. Primer sequences used to detect murine IL-1 and
GAPDH are shown in Table 1. Thermocycling conditions were as follows: 95 °C
for 1 min, 35 cycles at 95 °C, denaturation for 5 s, then annealing at 60 °C for 1 min. The level of GAPDH was employed as RNA content loading control.
Data were analyzed using the CT-method (2).
Table 1.The primers for rat IL-1 and GAPDH.
| Gene |
Primers |
Sequences (5′-3′) |
| IL-1 |
Forward |
TTCTTGGGACTGATGT |
| Reverse |
GAATGACTCTGGCTTTG |
| GAPDH |
Forward |
TTCAATGGCACAGTCAAGGC |
| Reverse |
TCACCCCATTTGATGTTAGCG |
2.3.7 ELISA for IL-1
Mouse serum, heart homogenates, or cell culture supernatant samples (100
L) were used to measure IL-1 using an ELISA kit. The analytical
sensitivity of the kit was 12.0 pg/mL and has a range of 31.3–2000 pg/mL,
respectively.
2.3.8 Western Blots
Tissue and cell samples were ground with RIPA buffer, and the mixture was
centrifuged at 12,000 g for 15 min at 4 °C. Supernatants were stored at –20 °C, and
the protein concentration was measured using a bicinchoninic acid (BCA) protein
assay kit (Beyotime Institute of Biotechnology). In total, 100 g of
proteins were separated using 5%–12% sodium dodecyl sulfate polyacrylamide gel
electrophoresis (SDS-PAGE). The proteins were transferred to PVDF membranes.
Following transfer, the membranes were treated with blocking buffer (5% BSA) for
2 h and subsequently incubated with primary antibodies against the
NF-Bp65s subunit (1:1000), phospho-NF-B (1:1000),
-arrestin1/2 (1:1000), NLRP3 (1:1000), IL-1 (1:1000), or GAPDH
(1:5000) at 4 °C overnight. After washing three times for 10 min with washing
buffer, the membranes were incubated with anti-rabbit IgG (1:1000) at room
temperature for 2 h. After another three washes for 10 min with washing buffer,
the protein bands were scanned and quantified using Image J software.
2.3.9 Statistical Analysis
Prism GraphPad software (version 8.0) (San Diego, USA) was used for all
statistical analyses. In each biological experiment, the data for each individual
western blot was normalized to a value of density in control (setting all control
values to 1). For control group, the data was normalized to the average value
(setting average values to 1) of biological replicates in order to perform the
statistical analysis. Data are presented as mean S.E.M. Comparisons were
conducted using one or two-way analysis of variance (ANOVA) followed by
Bonferroni’s post hoc test. p 0.05 was considered statistically
significant.
3. Results
3.1 AVP Induces Expression of the NLRP3 Inflammasome and
IL-1 in Mouse Myocardium
To determine if AVP perfusion induced expression of the NLRP3 inflammasome and
IL-1 in mouse myocardium, mice were injected with 2 g/g body
weight of AVP or 0.9% of NaCl, as a control, via the tail vein. Mouse hearts
were cut for frozen sections 6 h post AVP dosing, and we subsequently performed
immunohistochemistry as shown in Fig. 1A–B. Fluorescent optical density was
determined using Image J. As shown in Fig. 1C,D, the expression of the NLRP3
inflammasome and IL-1 protein in heart tissue were significantly
increased following AVP injection when compared with controls.
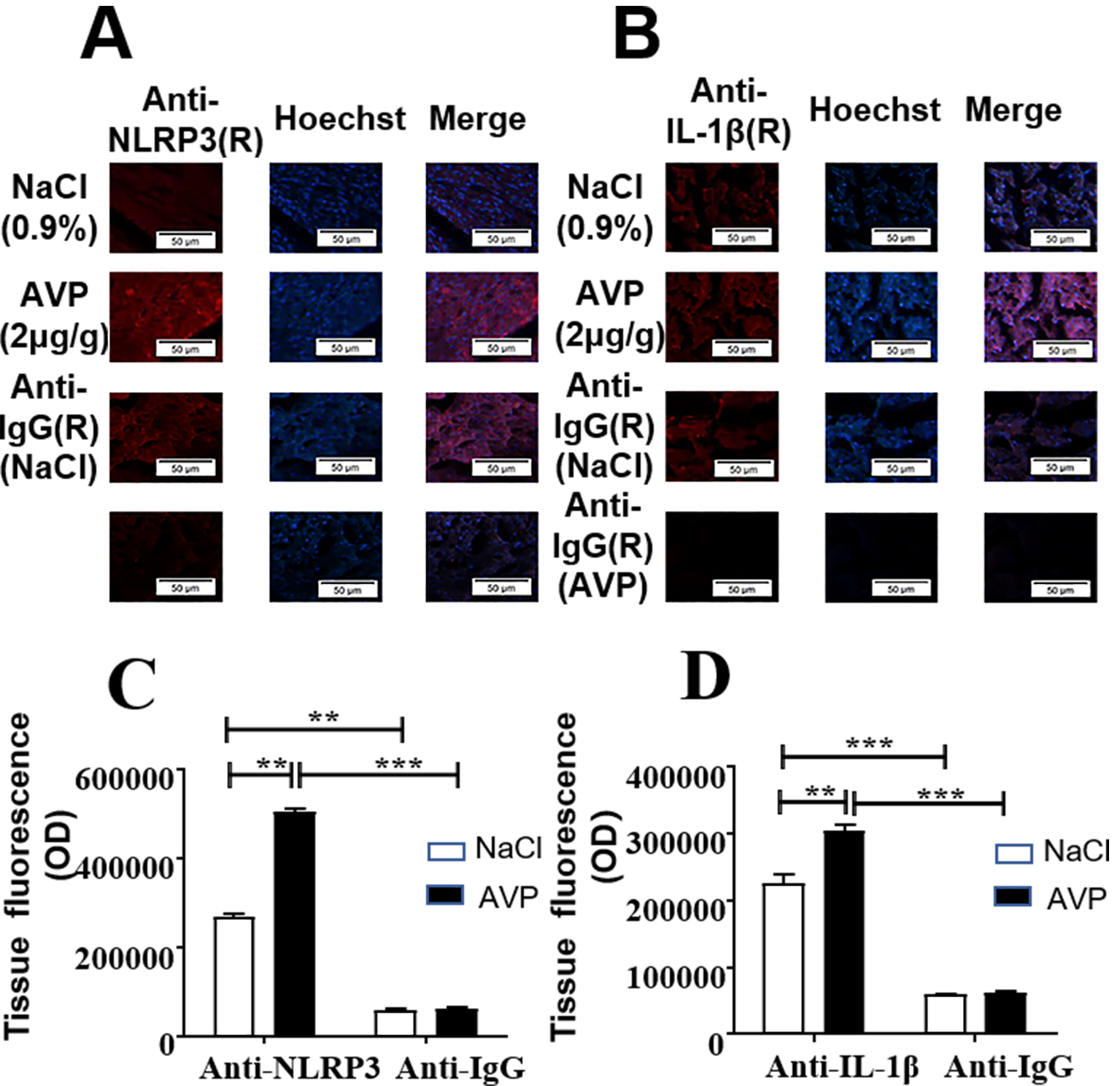 Fig. 1.
Fig. 1.
AVP induced expression of the NLRP3 inflammasome and
IL-1 in mouse myocardium following AVP administration.
(A,B) Representative images of wild type mice injected with 2 g/g
body weight of AVP or 0.9% of NaCl via the tail vein. Frozen sections were
prepared from dissected heart tissue to determine the protein level of the NLRP3
inflammasome and IL-1 6 h after dosing with NaCl or AVP.
(C,D) Fluorescent optical density was determined and data expressed as
mean S.E.M. of 3 separate animals with triplicate tissue sections,
**p 0.01, ***p 0.001. one-way ANOVA test, followed by
Bonferroni’s post hoc test. Note: Non-specific binding staining was examined
using mouse IgG. Scale bar = 50 m.
3.2 AVP Specifically Induced Expression of IL-1 in ARCFs
but not in ARCMs
To investigate whether AVP affected the viability of ARCMs, in vitroexperiments using ARCMs were performed. The cells were incubated with 10 M
AVP for 0 h, 12 h, 24 h, and 36 h followed by Hoechst/PI staining, and the number
of stained cells was counted under fluorescence microscopy. The viability of
cells was below 10% within 36 h of culture, indicating that AVP did not induce
the cell death (data not shown). To further investigate the effect of AVP on
IL-1 expression in ARCMs, primary rat cardiomyocytes were stimulated
with 10 M AVP for 0 h, 3 h, 6 h, 12 h, 24 h, or 36 h, and total RNA and
protein were isolated from ARCMs. The levels of IL-1 mRNA measured by
qPCR did not change significantly in ARCMs (Fig. 2A). In addition, the results of
ELISA (Fig. 2B–D) and western blotting (Fig. 2C) revealed that AVP did not alter
the expression of either IL-1 mRNA or protein in ARCMs during these time
periods.
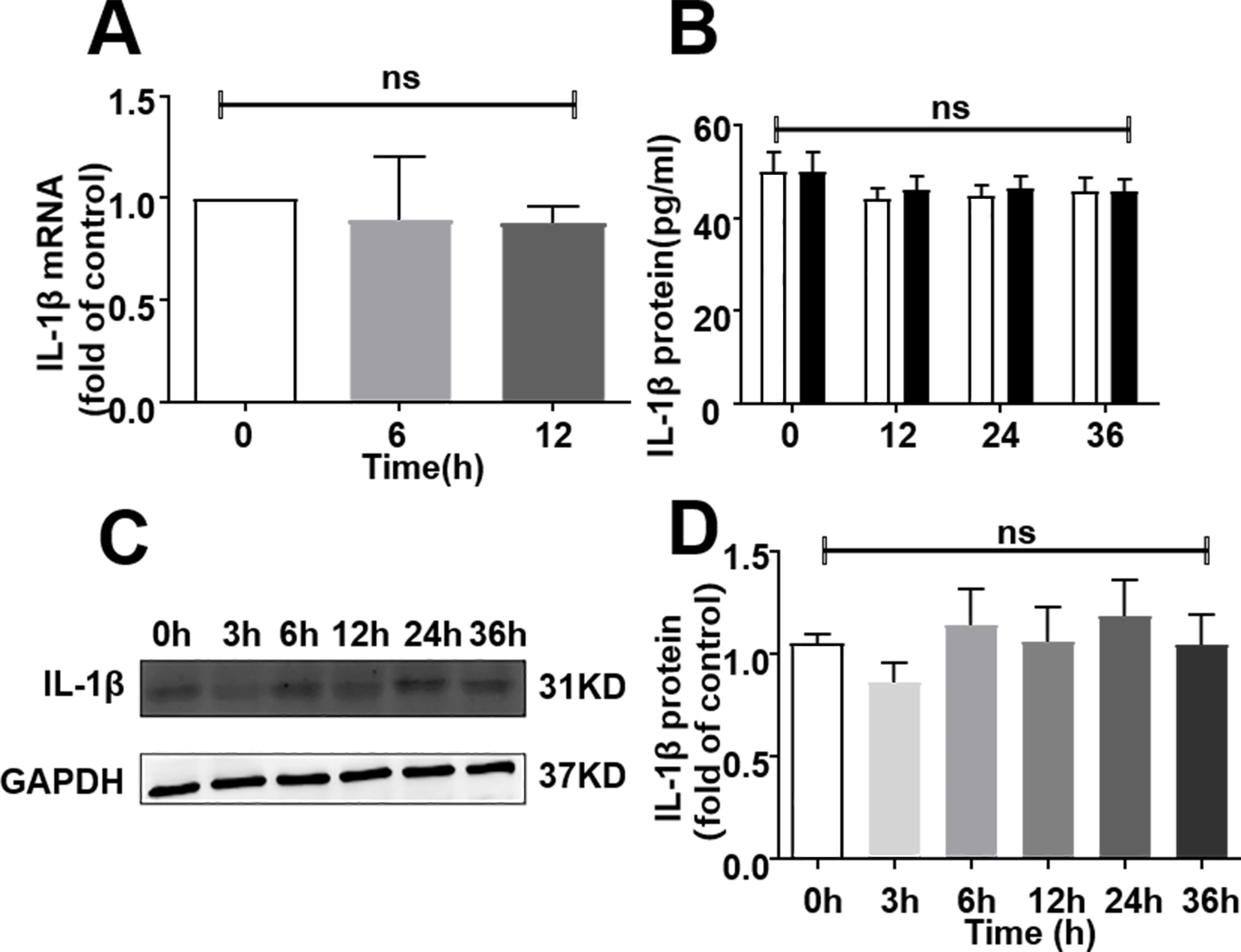 Fig. 2.
Fig. 2.
AVP did not induce IL-1 expression in adult rat
cardiomyocytes (ARCMs). (A) AVP treatment did not alter the level of
IL-1 mRNA in ARCMs. Cells were treated with 10 M AVP for 0–36 h
and subsequently harvested for total RNA. IL-1 mRNA was subsequently
determined by q-PCR. Data were expressed as mean S.E.M. of 4 separate
experiments. (B,C,D)AVP stimulation did not increase the level of
IL-1 protein in ARCMs. The cells in 6-well plates were stimulated with
10M AVP for 0–36 h. The supernatant was collected and ELISA assays
conducted to measure IL-1. Data are expressed as mean S.E.M. of
6 separate experiments. (B) Supernatant in cultured ARCMs.
(C,D) A represent blots and average data in ARCMs. One-way ANOVA test,
ns: no significance.
The same procedure was conducted with adult rat cardio fibroblasts (ARCFs) to
explore the effect of AVP on IL-1 induction. Starved ARCFs were
incubated with or without 10 M AVP for 0–24 h and cellular lysates were
harvested. The level of IL-1 in the supernatant of cultured medium was
detected by ELISA. Fig. 3A indicated that the incubation of ACRFs with AVP
increased the level of IL-1 protein in the supernatant of the cultured
ARCFs in a time-dependent manner compared with the vehicle control group.
However, the level of IL-1 protein in the cellular lysates determined by
western blot was not changed following stimulation, implying that mature
IL-1 were secreted from ARCFs, and the extra cellular IL-1
protein did not degrade within 24 h as shown in Fig. 3B,C.
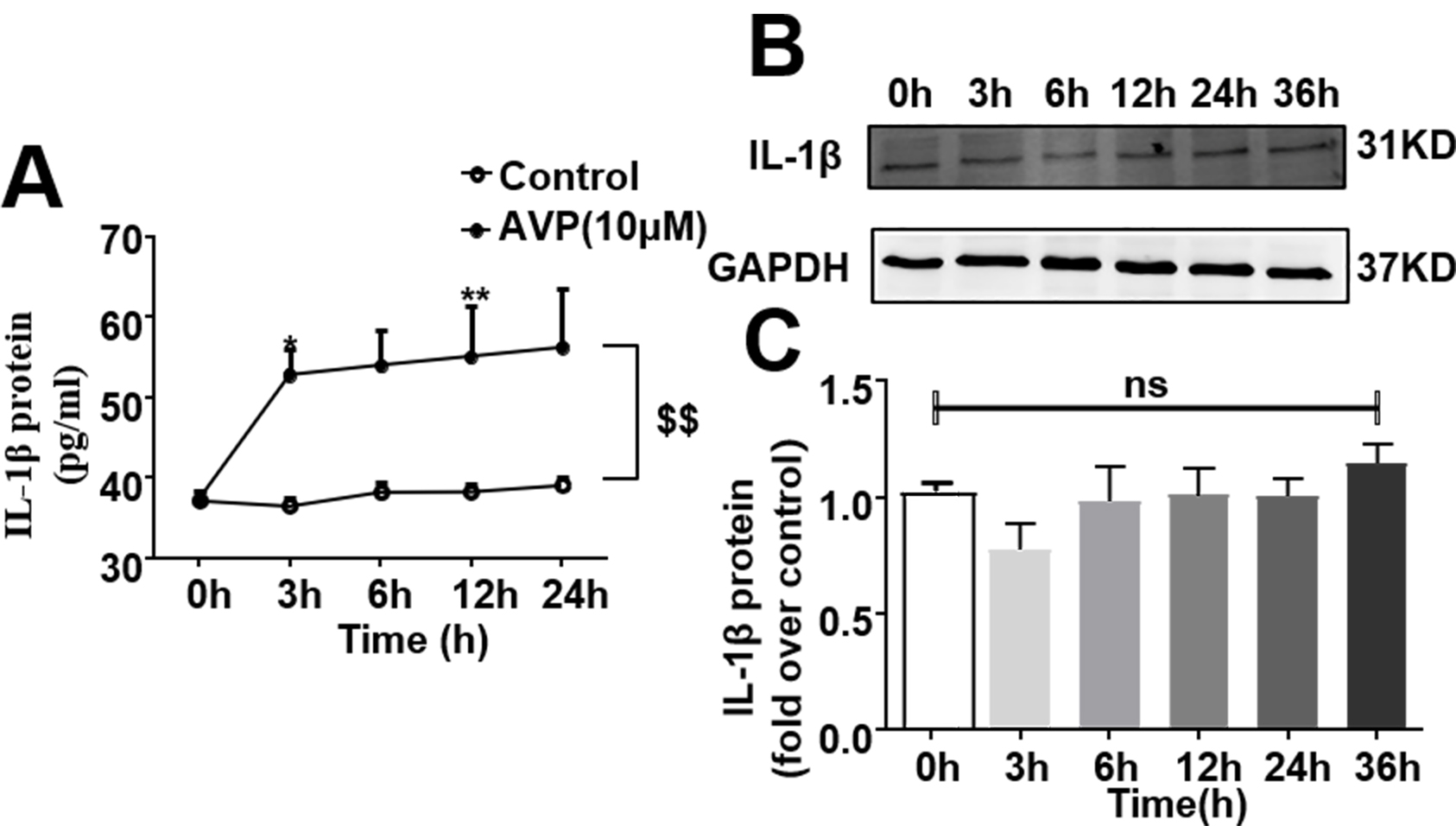 Fig. 3.
Fig. 3.
AVP induced the expression of IL-1 protein in adult rat
cardiac fibroblasts (ARFCs) in a time-dependent manner. Starved cells in 6-well
plates were stimulated with 10 M AVP for 0–24 h. (A) AVP increased
the level of IL-1 protein in a time-dependent manner in supernatant of
cultured cells. IL-1 was measured in culture media using ELISA. Data
were expressed as mean S.E.M. of 6 separate experiments. p 0.05, p 0.01, p 0.01 for
time-course (repeated two-way ANOVA). (B,C) AVP did not enhance the
IL-1 expression in the cellular lysates of ARCFs by either western
blotting or ELISA. Data were expressed as mean S.E.M. of 3 separate
experiments. One-way ANOVA test, ns, no significance.
3.3 AVP Induced the Expression of NLRP3 Inflammasome and
IL-1 through NF-B Signaling Pathway
To determine whether AVP induced NF-B p65 phosphorylation and whether
this process can be specifically blocked by PDTC, an inhibitor of NF-B,
starved ARCFs were pretreated with 50 M of PDTC followed by 10 M
AVP. The results showed that AVP induced NF-B p65 phosphorylation,
while PDTC specifically blocked AVP-induced this phosphorylation event (Fig. 4A,B). Moreover, PDTC itself had no effect on NF-B p65 phosphorylation.
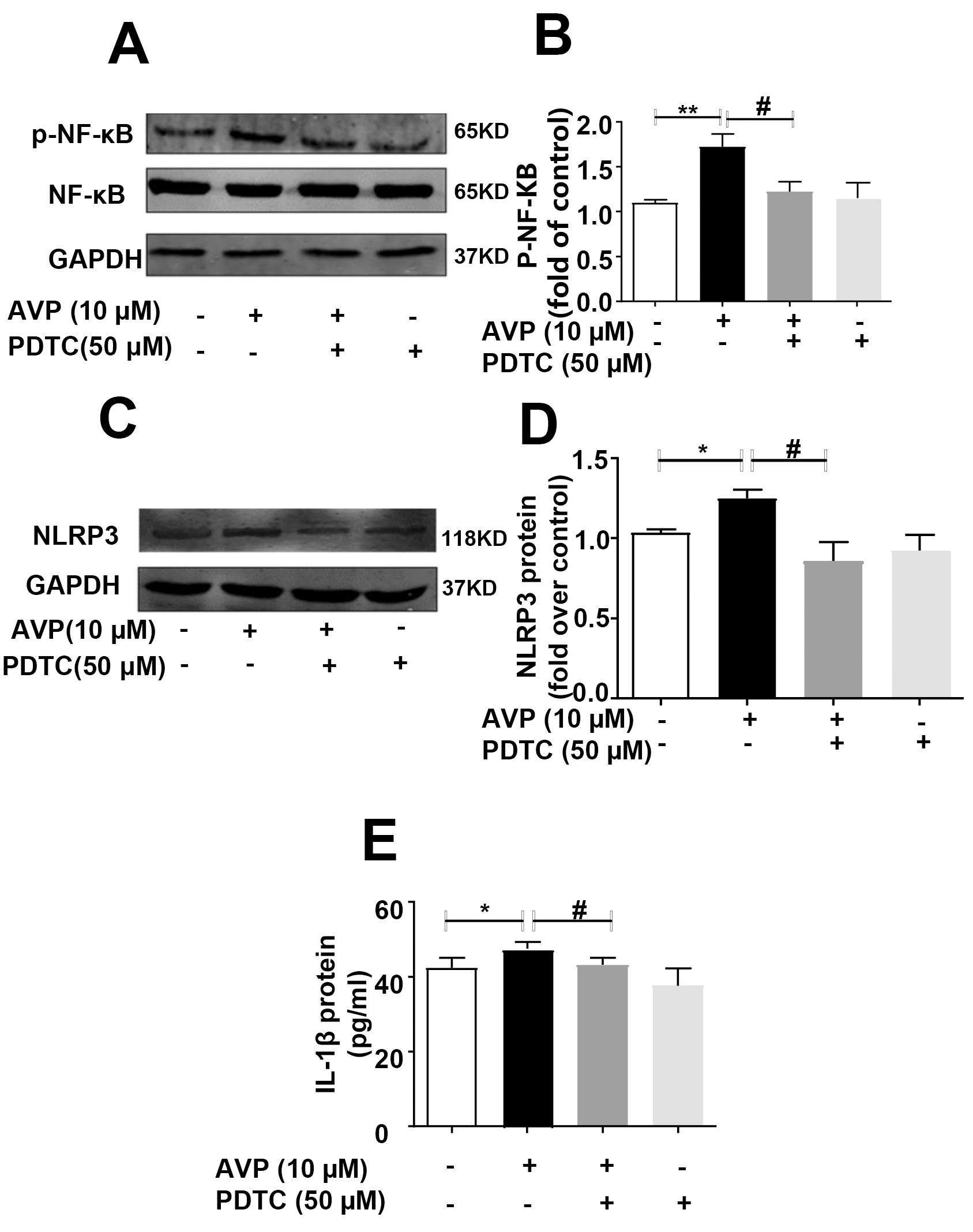 Fig. 4.
Fig. 4.
PDTC abolished AVP-induced the phosphorylation of NF-B
p65, expression of NLRP3 inflammation, and IL-1 in ARCFs. Starved cells
were cultured with 50M PDTC for 1 h, followed by stimulation with
10 M of AVP for 1 h. Cellular lysates were subsequently used in western
blotting experiments. (A) Representative blot. (B) Average data.
Data were expressed as mean S.E.M. of 6 separate experiments.
p 0.01, p 0.05, one-way ANOVA test,
followed by Bonferroni’s post hoc test. (C) PDTC abolished the
AVP-induced production of IL-1 protein in supernatant of cultured ARFCs.
(D,E) PDTC abolished the AVP-induced expression of NLRP3 inflammasome in
ARFCs. (D) A representative blot, (E): average data from 6 separate
experiments. *p 0.05, #p 0.05, one-way ANOVA test,
followed by Bonferroni’s post hoc test.
Accordingly, as displayed in Fig. 4C–E, the inhibition of NF-B with
PDTC6, significantly attenuated the effects of AVP in the induction of NLRP3 in
ARCFS, as well as enhancing the level of IL-1 in the supernatant of
cultured ARCFs compared to controls. Additionally, PDTC itself had no effect on
the expression of the NLRP3 inflammasome and IL-1.
3.4 The Silencing of -arrestin2 Prevented NLRP3 Expression
Induced by AVP Stimulation in ARCFs
-arrestin2 was knocked down in ARCFs using shRNA-encoding lentivirus.
48 h post infection, more than 90% of the cells were infected as evidenced by
cells displaying virally-encoded fluorescent reporter (data not shown).
Subsequently, western blotting was used to determine that either shRNA898 or
shRNA900 significantly down regulated the expression of -arrestin2
compared to a scrambled shRNA control, as shown in Fig. 5A,B. In ARCFs with and
without knocked down -arrestin2 expression, western blot analysis
indicated that AVP induced the expression of the NLRP3 inflammasome in ARCFs, but
not in ACRFs with knocked down -arrestin2 expression (Fig. 5C,D). This
suggested that silencing -arrestin2 abolished AVP-induced expression of
NLRP3 in ARCFs.
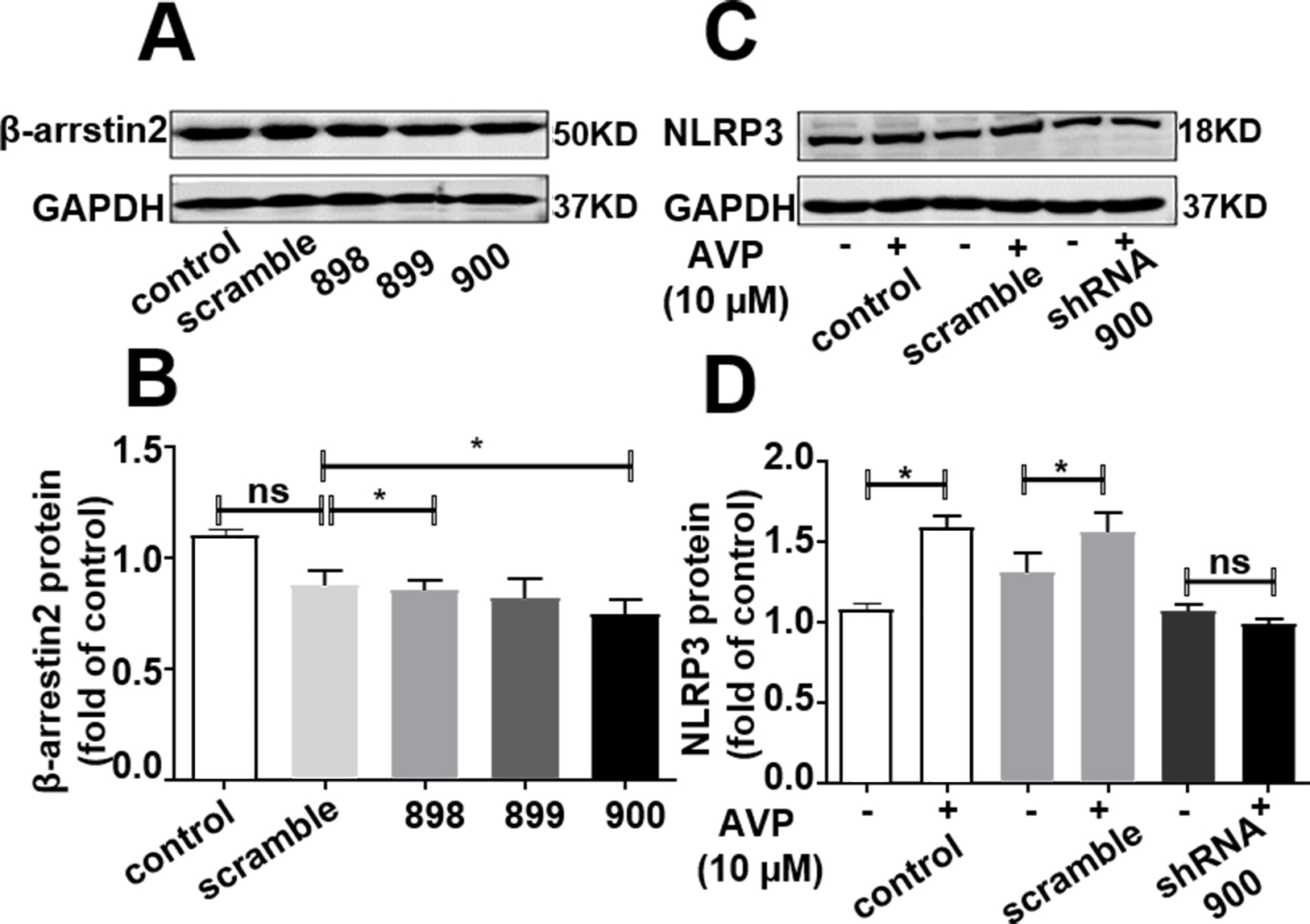 Fig. 5.
Fig. 5.
Silencing of -arrestin2 abolished NLRP3 expression
induced by AVP stimulation in ARCFs. (A,B) Silencing of
-arrestin2 protein expression by different -arrestin2 shRNA in
ARCFs. Western blotting confirmed decreased the expression of -arrestin2
in ARCFs. (A) A representative blot showing that shRNA-encoding
lentivirus efficiently knocked down expression of -arrestin2.
(B) Average data. Data were presented as the mean S.E.M. from 6
independent experiments. p 0.05 vs scramble, one-way ANOVA
test, followed by Bonferroni’s post hoc test. (C,D) Knockdown of
-arrestin2 abolished the AVP-induced NLRP3 inflammation expression in
ARCFs. (C) A representative blot. (D) Average data. Data were
presented as the mean S.E.M. from 5 independent experiments.
p 0.05, one-way ANOVA test, followed by Bonferroni’s post hoc
test. ns, no significance.
3.5 -arrestin2 was Necessary for AVP-Induced IL-1
and NLRP3 Inflammasome Expression in Mouse Myocardium
To investigate the specific role of -arrestin2 in AVP-induced
inflammatory response, -arrestin2 knockout mice and wild-type mice were
injected with AVP (2 g/g body weight) through the tail vein. An equal
volume of normal saline with 0.9% NaCl was injected into the vehicle group. The
protein level of IL-1 in blood serum, as measured by ELISA, indicated
that dosing of AVP enhanced the level of IL-1 in the serum of wild type
mice, but not in -arrestin2 knockout mice (Fig. 6A). Further,
IL- protein expression of in the left ventricle of mouse heart measured
by ELISA (Fig. 6B) and western blot (Fig. 6C,D) demonstrated that AVP induced the
expression of IL-1 in myocardium of wild type mice, but not in
-arrestin2 knockout mice. These results suggest that AVP-induced
IL-1 expression in cardiac tissue is dependent on -arrestin2.
Similarly, as shown in Figs. 6E,6F, dosing of AVP induced the expression of the
NLRP3 inflammasome in the heart tissue of normal mice, but not in
-arrestin2 knockout mice. Therefore, AVP-induced NLRP3 inflammasome
expression dependent on -arrestin2 signaling.
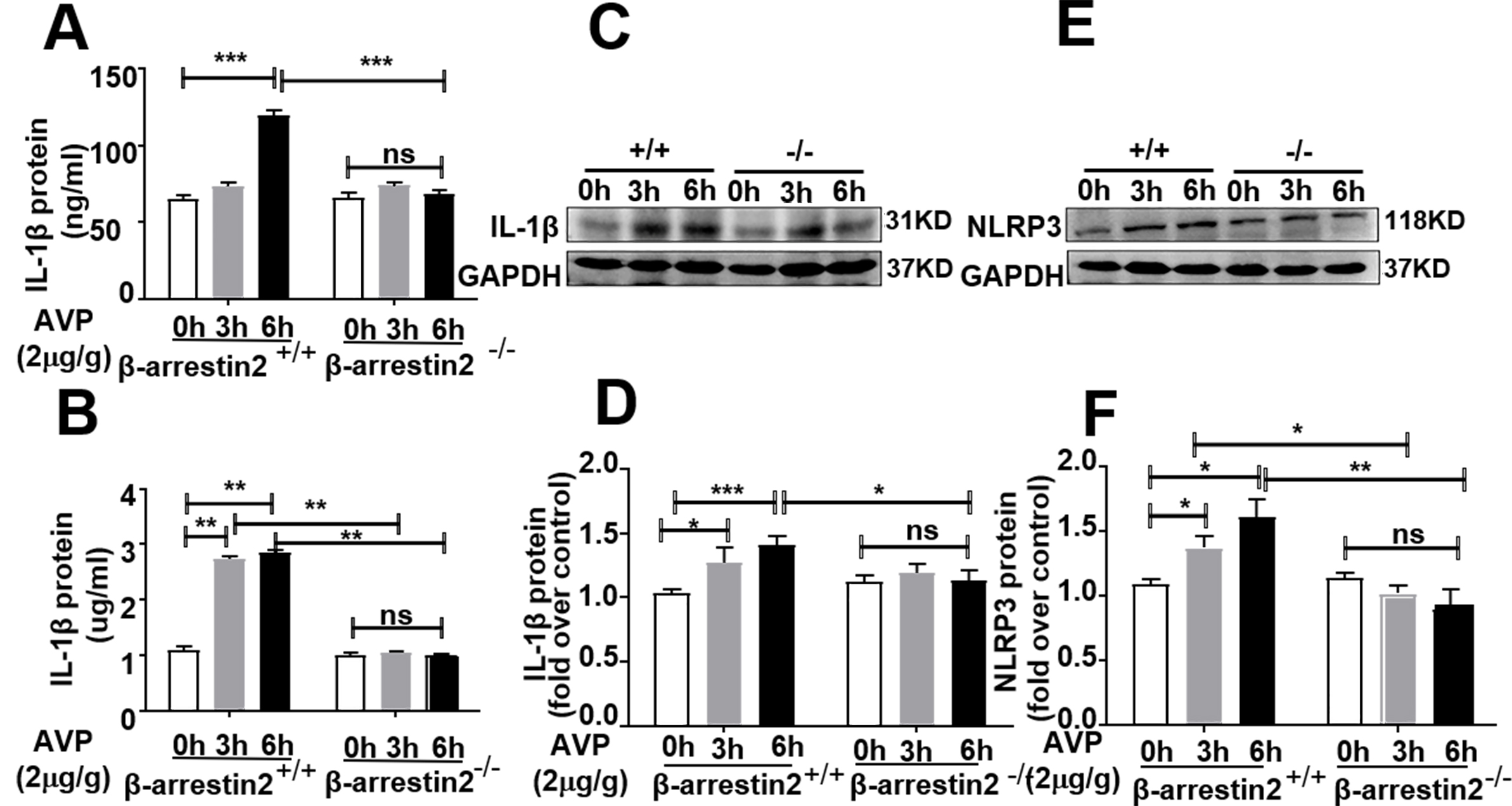 Fig. 6.
Fig. 6.
-arrestin2 was necessary for AVP-induced IL-1
expression in mouse myocardium. (A) AVP induced the expression of
IL-1 protein in serum, but not in -arrestin2 knockout mice.
Lysates from the left ventricle and serum were harvested 0–6 h post injection
with 2 g/g of AVP or 0.9% of NaCl into tail veins. The serum was
collected for measurement of IL-1 as described in Methods. Data were
expressed as mean S.E.M. of 5 separate experiments. p 0.001 vs control, one-way ANOVA test, followed by Bonferroni’s post hoc
test. (B,C,D) Deletion of -arrestin2 abolished AVP-induced
IL-1 expression in mouse myocardium. (B) Content of
IL-1 in mouse myocardium. (C) A representative blot.
(D) Average data. Data were expressed as mean S.E.M. of 4
separate animals. p 0.05, p 0.001,
one-way ANOVA test, followed by Bonferroni’s post hoc test.
(E,F) Knockout of -arrestin2 abolished AVP-induced NLRP3
expression. (E) A representative blot. (F) Average data. Data
were expressed as mean S.E.M. of 6 separate animals. p
0.05, p 0.01. One-way ANOVA test, followed by Bonferroni’s
post hoc test. ns, no significance.
3.6 -arrestin2 is Involved in AVP-Induced Activation of the
NF-B Signaling Pathway in Mouse Myocardium
To identify the mechanism mediating the effect of -arrestin2 on
AVP-induced expression of IL-1 and the NLRP3 inflammasome, mice were
treated as mentioned in 2.5. Western blot analysis showed that AVP induced
NF-B p65 phosphorylation in the heart tissue of wild-type mice compared
to controls. However, AVP had no effect on NF-B p65 phosphorylation in
-arrestin2 knockout mice (Fig.7A,B). In addition, the deletion of
-arrestin2 inhibited the basal level of NF-B p65
phosphorylation.
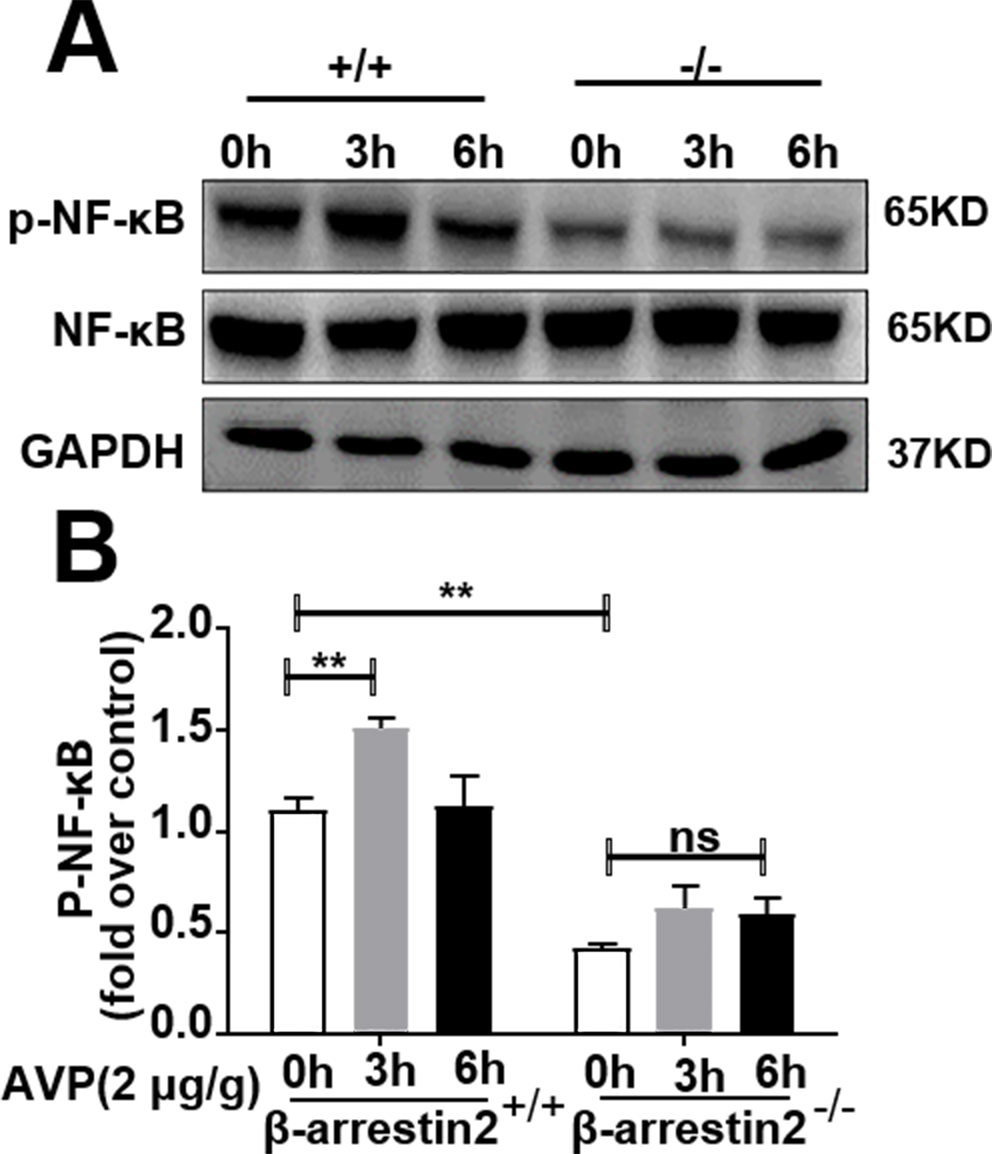 Fig. 7.
Fig. 7.
Deletion of -arrestin2 diminished phosphorylation of
NF-B p65 induced by AVP in mouse myocardium. Left ventricle were
harvested 0–6 h post injecting 2 g/g body weight of AVP or 0.9% of NaCl
into veins of the tails. The myocardium lysate was subsequently assayed for
phosphorylation of NF-B p65 with anti-phospho NF-B p65
antibody by western blotting. (A) A representative blot.
(B) Average data were from 3 individual animals, p
0.05, p 0.01, one-way ANOVA test, followed by Bonferroni’s
post hoc test. Note: -arrestin2 KO reduced the basal level of
NF-B p65 phosphorylation.
4. Discussion
Arginine vasopressin (AVP) is a mammalian vasopressin peptide synthesized mainly
in neuroendocrine neurons in the supraoptic, paraventricular, and suprachiasmatic
nuclei of the hypothalamus [15]. A number of experimental and clinical studies
have shown that the concentration of AVP in the blood of patients with
hypertension and post-infarct heart failure is significantly increased. Further,
increased levels of inflammatory cytokines (TNF-, IL-1, IL-6)
[16], and high levels of AVP can lead to heart failure [17]. Early studies have
reported that AVP induces the expression of IL-6 in cardiac fibroblasts, promotes
the proliferation of neonatal rat AVP in cardiac fibroblasts [6], and this leads
to myocardial fibrosis [18]. The present study found that: (1) AVP induces
expression of the NLRP3 inflammasome and IL-1 in mouse heart and
cultured rat cardio fibroblasts; (2) AVP-induced expression of IL-1 in
adult cardiac fibroblasts but not in adult rat cardiomyocytes; (3) AVP induces
the expression of NLRP3 inflammasome and IL-1 through the NF-B
signaling pathway; (4) AVP-induced expression of IL-1 and activation of
NF-B signaling is dependent on -arrestin2. Therefore, AVP
promotes the expression of IL-1 through the -arrestin2-mediated
NF-B signaling pathway, and participates in the regulation of
inflammation.
Fibroblasts are traditionally considered extracellular matrix-producing cells
that become activated following injury and participate in scar formation [19].
Nevertheless, role of cardiac fibroblasts in promoting inflammation have
attracted more attention recently. Cardiomyocytes [20], cardiac endothelial cells
[21], cardiac fibroblasts [6, 22, 23], and cardiac infiltrates [24, 25] were all
reported to produce inflammatory cytokines that cause cardiac remodeling and
dysfunction. Among them, IL-1 mainly comes from cardiac endothelial
cells and cardiac fibroblasts and is strictly regulated by inflammasomes [26, 27]. Cardiac fibroblasts are particularly sensitive to the pro-inflammatory
cytokine IL-1 in post-myocardial infarction remodeling [28]. IL-1 is involved in
the pathogenesis of a variety of inflammatory diseases, in which both
IL-1 and IL-18 are generally not expressed in healthy cells, but their
inactive precursors can be synthesized after cell injury and both molecules are
secreted as active molecules. The present study used ARCMs and ARCFs as the
experimental subjects. Interestingly, the expression of IL-1 could not
be detected in ARCMs, but the levels of IL-1 and NLRP3 inflammasome
expression were increased in ARCFs. This was confirmed in another study that
showed that isoproterenol induced the expression of IL-18, but not IL-1,
in cardiomyocytes [29]. The difference of reactivity between the cardiac cell
types might provide experimental basis for drug selectivity in clinical practice.
It is well known that -arrestins are scaffold proteins involved in GPCR
desensitization and down-regulation, and can transduce receptor signals
independently of G-proteins. A recent study reported that cardiac
-arrestin2 expression is increased in mice with myocardial infarction
and modulates the inflammatory response [30]. Additionally, earlier work in our
laboratory revealed that the -arrestin1 and -arrestin2 pathways
are involved in AVP-induced cardiac fibroblast proliferation and extracellular
matrix synthesis, and that -arrestin2 is also involved in AVP-induced
fibroblast IL-6 expression. This study used -arrestin2 knockout mice and
-arrestin2 knockdown ARCFs as experimental models. The effects of
-arrestin2 on AVP-induced expression of the NLRP3 inflammasome and
IL-1 were investigated in both animal and cellular models. A highlight
of this study is the discovery of the relationship between -arrestin2
and the NLRP3 inflammasome during cardiac inflammation. Specifically, we document
that -arrestin2 is a necessary molecule for the AVP-induced expression
of the NLRP3 inflammasome.
An involvement of the NLRP3 inflammasome has been shown in cardiovascular
diseases. A few pharmacological agents that selectively target the NLRP3
inflammasome signaling have been tested in animals, and early phase human studies
show promising results [31, 32]. It is well known that there are different
inflammasomes that are comprised of a complex of proteins. They activate
caspase-1 which, in turn, promote maturation of pro-IL-1 and IL-18 into their
active forms. Among the different types of inflammasomes such as NLRP1, NLRP2,
NLRP3 and AIM, the role and regulation of Nodlike receptor family, pyrin domain
containing 3 (NLRP3) inflammasome is well studied [33]. Such as in the seminal
plasma of varicocele patients, NLRP3 inflammasome components include
interleukin-1b (IL-1b), IL-18, caspase-1, and apoptosis associated speck-like
protein (ASC) [34]. Non-NLRP3 inflammasomes may be significant in cardiovascular
diseases as well [35, 36]. It is worthwhile to further study if other components
are involved in AVP-induced NLRP3 inflammasome and non-NLRP3 inflammasome
signaling beyond the IL-1, although our lab reported that IL-6 mediates
AVP-evoked the cardiac inflammation [6].
It is well known that NF-B is an important nuclear transcription
factor that induces inflammatory signaling and the expression of inflammatory
factors. Further, NF-B has been proved to be involved in the occurrence
and development of various immune and inflammatory responses, apoptosis, and
proliferation-related heart diseases [37]. It has been shown that the
TLR4/MyD88/NF-B signaling pathway is involved in myocardial
inflammation after coronary microembolization, evidenced by increased serum
levels of TNF-, IL-1, and increased expression levels of
TNF-, IL-1 and IL-18 in myocardium. Among these, TLR4 was
described as the first member of the TLR family, while MyD88 is a key adapter
molecule in the Toll-like receptor signaling pathway, connecting the activation
of NF-B and the NLRP3 inflammasome [38]. The present study indicated
that the NF-B signaling pathway was involved in the arginine
vasopressin-induced expression of the NLRP3 inflammasome and IL-1 in vivo and in vitro. We further showed that PDTC, an inhibitor
of NF-B, inhibited the inflammatory response to some extent by
inhibiting the phosphorylation of NF-B p65. In addition, knockout of
-arrestin2 proved to inhibit NF-B 65 phosphorylation as well.
The regulatory effect of NF-B suggests that it may provide insights
into inflammation in cardiovascular disease.
5. Conclusions
In conclusion, this study shows that AVP induces the expression of IL-1
specifically in ARCFs, and -arestin2 is necessary for the activation of
the arginine vasopressin-induced NF-B signaling pathway. Therefore,
silencing of -arrestin2, abolishing NF-B signaling, and
inhibiting the NLRP3 inflammasome all provide a novel approach for the clinical
treatment of inflammatory cardiovascular diseases.
Abbreviations
AVP, Vasopressin; ARCFs, adult rat cardiac fibroblasts; ARCMs, adult rat
cardiomyocyte; NF-B, Nuclear factor-Kappa B; GAPDH,
Glyceraldehyde-3-phosphate dehydrogenase; IL, Interleukin; TNF, Tumor Necrosis
Factor; ELISA, Enzyme-linked immunosorbent assay.
Availability of Data and Materials
The datasets used and/or analyzed during the current study are available from
the corresponding author on reasonable request.
Author Contributions
NY, BG, and WZ designed the research study. NY, BG, YW, YH, XZ, JC, YL, YQ, and HS performed the research. NY, BG, and WZ analyzed the dat. NY, BG, and WZ wrote the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Ethics Approval and Consent to Participate
The study was approved by the Board of Nantong University Animal Care and Use (Ethical approval number: NTU220191418).
Acknowledgment
Warmly thank Dr. Lefkowitz (Duke University) for kindly providing
-arrestin2 knock out mice and Ms. Emily Zhu (University of Virginia) for
a critical reading of the manuscript.
Funding
The study was financially supported by National Natural Science Foundation of
China (No. 81770400 to WZ) and Nantong Municipal Science and Technology
(JCZ18131&JC2019133 to HS).
Conflict of Interest
WZ is serving as one of the Guest Editors of this journal. We declare that WZ had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial
process for this article was delegated to SI and MMA.









