
†These authors contributed equally.
Academic Editor: Graham Pawelec
Background: As a fatal cardiovascular complication,
coronary microembolization (CME) results in severe cardiac dysfunction and
arrhythmia associated with myocardial inflammation and apoptosis. Human urinary
kallidinogenase (HUK) can provide a protective function for cardiomyocytes by
improving microcirculation. However, the therapeutic effects and underlying
mechanisms of HUK in CME-induced myocardial injury remain unclear.
Aims: We evaluated the effect of HUK on cardiac protection in a
rat model of CME and whether it could restrain myocardial inflammation and
apoptosis, and alleviate CME-induced myocardial injury. Methods: We
established the CME model by injecting 42
Coronary artery microembolization (CME) is a major cause of “no recurrent flow” in occluded coronary arteries after interventional procedures [1, 2]. It is considered to be the main complication of percutaneous coronary intervention (PCI) [3]. CME may disrupt myocardial reflow, leading to severe cardiac dysfunction and arrhythmia [4]. Moreover, a study by Heusch et al. [5]indicated that repetitive CME could cause progressive loss of functional cardiomyocytes and induce heart failure. Rat CME has also been reported to display a local myocardial infarction, with a large number of apoptotic and necrotic cells in the corresponding area [6]. Therefore, existing studies indicate that myocardial inflammation and apoptosis may play vital roles in CME-induced myocardial damage, and targeted interventions in these aspects might be critical for myocardial protection [7, 8, 9].
Recently, the potential protective mechanism of the PI3K/Akt axis in CME has received considerable attention. For example, gustazine has been shown to inhibit oxidative stress and inflammation by influencing the PI3K/Akt pathway, thereby reducing myocardial damage caused by CME in rats [10]. Human urinary kallidinogenase (HUK), a glycoprotein found in male urine that modulates the kallikrein-kinin system, has been used in a wide range of patients diagnosed with ischemic stroke [11]. Ma et al. [12] confirmed that HUK acts as an anti-inflammatory and anti-apoptotic factor through the PI3K/Akt/FoxO1 signaling pathway, thereby reducing brain damage in rats with persistent middle cerebral artery occlusion. However, further research is required to confirm whether HUK can reduce myocardial damage caused by CME and improve cardiac function through the PI3K/Akt/FoxO1 signaling pathway. This study aimed to explore the effect of HUK on myocardial inflammation and apoptosis and its role in the PI3K/Akt/FoxO1 pathway to determine whether it improves CME-induced cardiac dysfunction and increases cardiac output.
The flow of our study was represented in Fig. 1. All experimental animals were
procured from the Experimental Animal Center of Guangxi Medical University. All
experimental protocols and steps according to the NIH (National Institutes of
Health) Guidelines for the Use of Laboratory Animals, and had the approval of the
Animal Ethics Committee. Firstly, 40 male adult Sprague-Dawley (SD) rats of
250–300 g were randomly divided into CME, CME + HUK treatment, CME + HUK + LY, and
sham operation groups, with ten rats in each group. We then built a CME model
based on the previously reported process [9]. Briefly, an intraperitoneal
injection of pentobarbital sodium (30–40 mg/kg) was administered to anesthetize
the SD rats. We used a small-animal ventilator to assist with breathing. We
conducted a left-sided thoracotomy in the incision position between the third and
fourth ribs of the rat. The ascending aorta was separated and gripped for 10 s.
Simultaneously, we mixed 3000 plastic microspheres of 42
 Fig. 1.
Fig. 1.The flow chart of our study.
Based on Chen et al. [4] study, 12 hours after modeling is the best time to measure cardiac function in rats while at the lowest level. At that stage, we evaluated the cardiac function of the rat using the Hewlett-Packard Sonos (Philips Sonos7500 system, Philips Technologies, USA) to measure the left ventricular fraction shortening (LVFS), left ventricular ejection fraction (LVEF), left ventricular end-systolic diameter (LVESd), and left ventricular end-diastolic diameter (LVEDd), respectively. The measurement was repeated thrice for each rat, and the average value was calculated. All the above operations were performed using an S12 probe with a 12 MHz probe. Experienced cardiac sonographers were used to perform all ultrasound examinations.
Blood samples were drawn from the abdominal aorta before sacrifice. The levels
of serum cardiac troponin I (cTnI), creatine kinase myocardial band isoenzyme
(CK-MB), lactate dehydrogenase (LDH), IL-1
Twelve hours after the operation, the rats were sacrificed for sampling after completing the ultrasound examination. We administered an intraperitoneal injection of pentobarbital sodium (60 mg/kg) to anesthetize rats. Blood was collected from the abdominal aorta and the heart was isolated shortly after opening the chest. The auricle and atrium were then excised, and the cardiac tissue was separated into upper, middle, and bottom sections. For the comparability of each group, the same inspection index was performed using the same part of cardiac tissue. The upper and middle sections were kept at –80 °C to prepare for real-time quantitative PCR (RT-qPCR) and western blotting. The bottom sections were immobilized in 4% paraformaldehyde for 12 h. It was then embedded in paraffin and used for slice preparation. After hematoxylin-eosin (H&E) staining, cardiomyocytes were observed after CME using an optical microscope. Hematoxylin-basic fuchsin-picric acid (HBFP) staining was used to identify myocardial ischemia, and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining was used to identify cardiomyocyte apoptosis.
Myocardial ischemia was detected via HBFP staining, which enabled the staining of ischemic cardiomyocytes and cytoplasm (red), and the nuclei of infarcted (yellow) and healthy (blue) cardiomyocytes. Each stained section was observed with a pathology image analyzer (DMR + Q550, Leica, Wetzlar, Germany). Five fields of view were obtained from each slice. Leica Qwin software (2.7, Leica Microsystems Inc, Buffalo Grove, IL, USA) and the planar method were used to calculate the infarct area. The infarct area was then divided by the entire observation area to measure the infarct percentage.
The TUNEL assay (Roche, USA) was performed using a TUNEL apoptosis detection kit
according to the manufacturer’s instructions to detect cardiomyocyte apoptosis.
The nuclei of apoptotic cells appeared yellowish-brown under light microscopy.
Forty nonoverlapping regions were randomly selected from each slice
(
We prepared slices of 4
Samples of heart tissue (200 mg) were processed by RT-qPCR. Following the manufacturer’s instructions, we extracted apoptosis-associated RNAs such as Bax, Bcl-2, and caspase-3, which were determined by RT-qPCR for relative levels in CME. The ABI PRISM-7500 machine (CA, USA) was used to perform RT-qPCR using the TB Green® Premix Ex Taq™ II (TaKaRa, Japan). The specific primer sequences used in this study were listed in Table 1.
| GENE | NCBI ID | Primer Sequence |
| Bcl-2 | 24224 | Forward: 5′-CCTGGCATCTTCTCCTTCCA-3′ |
| Reverse: 5′-GGACATCTCTGCAAAGTCGC-3′ | ||
| Bax | 24887 | Forward: 5′-AAGAAGCTGAGCGAGTGTCT-3′ |
| Reverse: 5′-CCAGTTGAAGTTGCCGTCTG-3′ | ||
| Caspase-3 | 25402 | Forward: 5′-TGTCGATGCAGCTAACCTCA-3′ |
| Reverse: 5′-GCAGTAGTCGCCTCTGAAGA-3′ | ||
| GAPDH | 24383 | Forward: 5′-TGTGAACGGATTTGGCCGTA-3′ |
| Reverse: 5′-GATGGTGATGGGTTTCCCGT-3′ |
Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) (stacking gel: 5% SDS-PAGE, separating gel: 10–15% SDS-PAGE) was utilized to separate the total protein obtained from cardiac tissues and then transferred it to a polyvinylidene fluoride membrane (PVDF, Millipore, Atlanta, Georgia, United States). The above experimental operations were all completed at a constant voltage of 100 V. The membrane was then sealed with BSA at 25 °C for 1 hour. Next, the membrane was hatched with corresponding primary antibodies at 4 °C for 24 hours. After 24 hours, the membrane was washed five times. We then incubated the secondary antibody (ab6721, 1:10000, Abcam) coupled to the enzyme for 2 hours.
All data were analyzed in SPSS software (version 23.0, IBM, Chicago, IL, USA). The results of our experiments are presented as mean standard deviation (SD). ANOVA was conducted to test the significance of differences in sample means, and a p-value
To assess circulatory changes in rats, we measured the LVEF, LVFS, LVEDd, and LVESd after 12 h in the CME model. The echocardiographic results of each group demonstrated that the CME group had lower LVEF and LVFS values than the Sham group, while LVEDd and LVESd were reversed (Fig. 2A,B). Additionally, cardiac function was improved in the CME + HUK group compared with the CME group, showing higher LVEF/LVFS and lower LVEDd/LVESd.
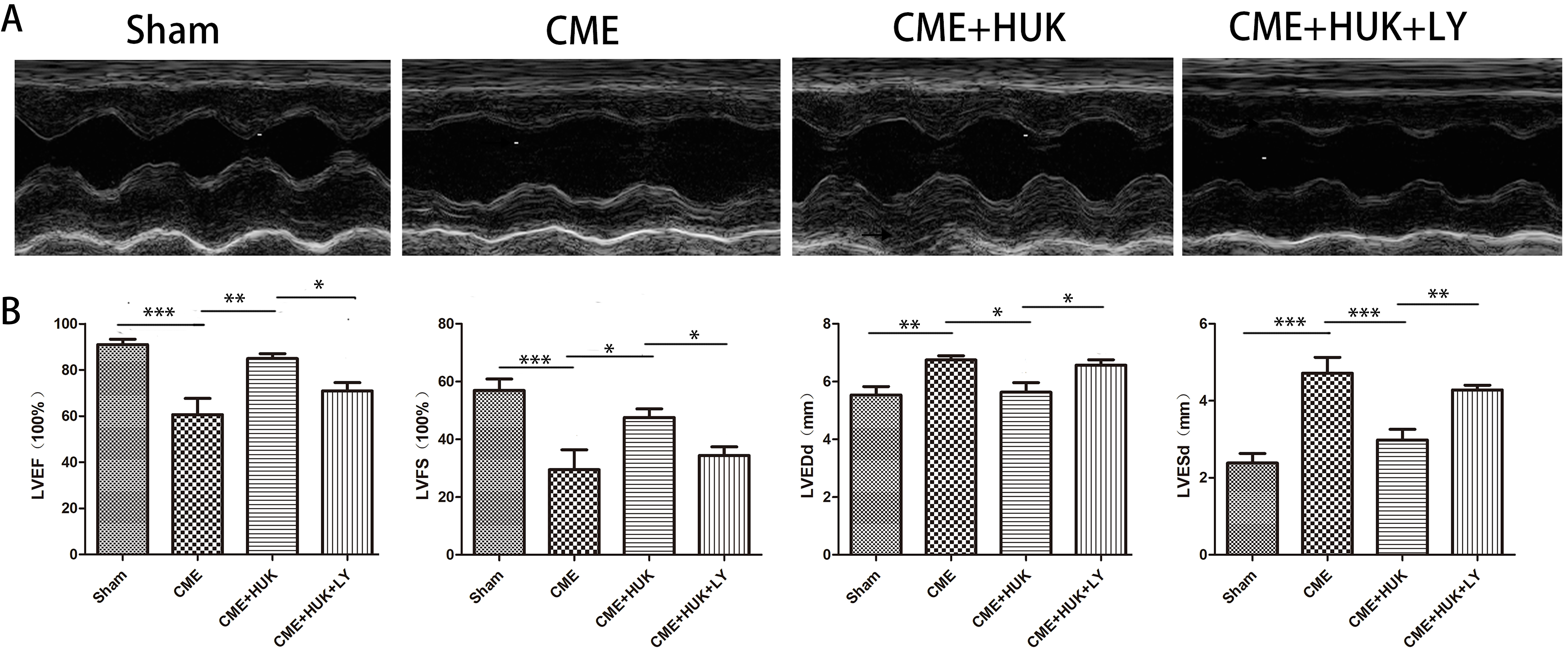 Fig. 2.
Fig. 2.Measurement of cardiac function. (A) Cardiac function evaluated
by echocardiography in each group (n = 6, per group). (B) The cardiac function
parameters of left ventricle ejection fraction (LVEF), left ventricle fractional
shortening (LVFS), left ventricular end-diastolic diameter (LVEDd), and left
ventricular end-systolic diameter (LVESd) were measured quantitatively. Data were
presented as the mean
H&E and HBFP staining suggested that there were sporadic ischemic areas in the
Sham group and most areas were normal filling tissues (Fig. 3A,B). In the CME,
CME + HUK, and CME + HUK + LY groups, micro-infarct areas were observed; most of
the infarct areas were located in the left ventricle, and embolized microspheres
could be seen in the field of vision. The results of H&E staining indicated that
cardiomyocytes in the micro-infarct area had no nuclei or ruptured nuclei; the
cytoplasm was also red. In addition, the area around the infarct showed swelling
and degeneration of cardiomyocytes, red blood cell effusion, and infiltration of
inflammatory cells. The HBFP staining results showed that the infarct area of CME
rats was significantly wider than that of the Sham group (p
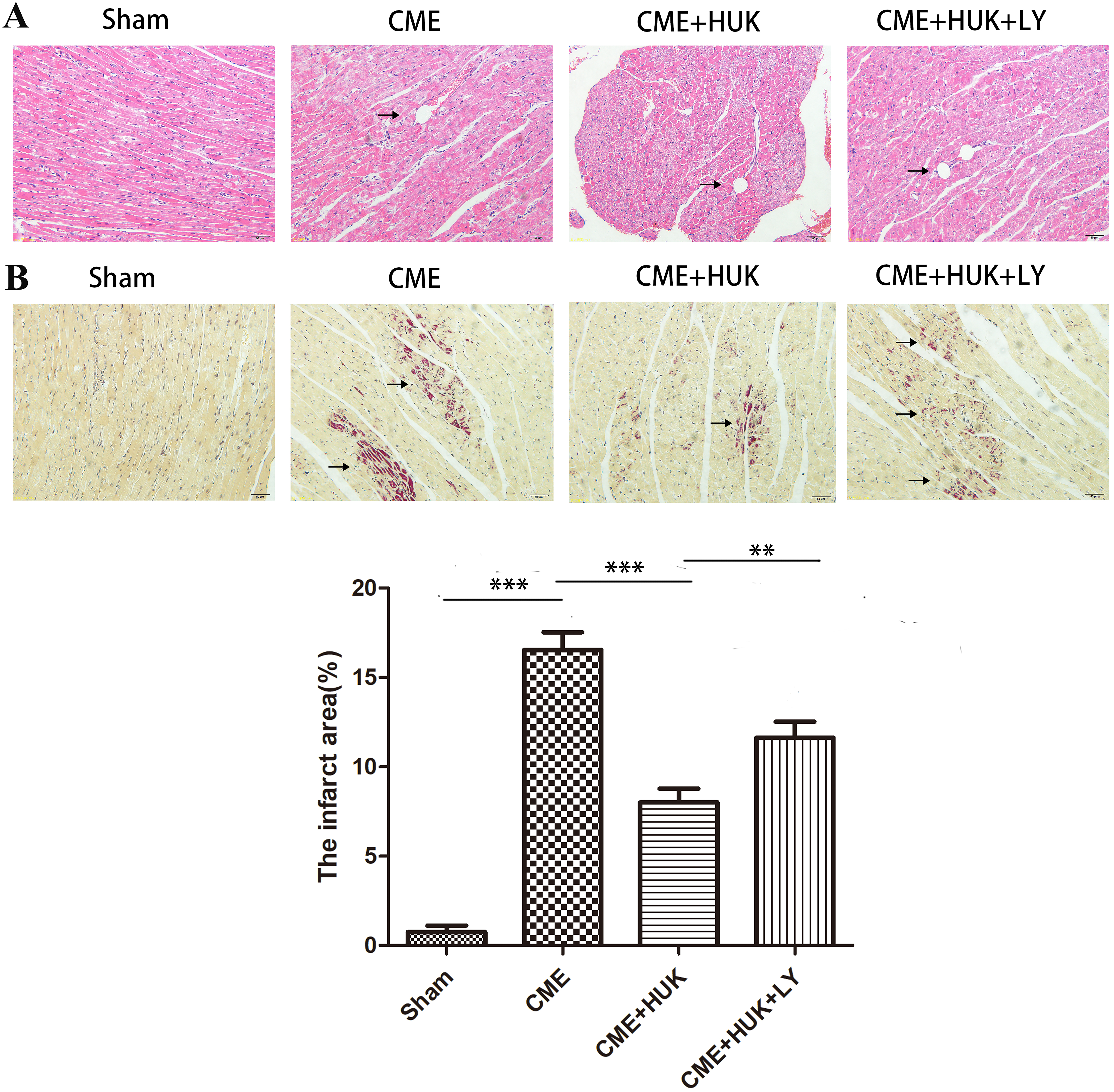 Fig. 3.
Fig. 3.H&E and HBFP staining of myocardial tissues (
TUNEL staining results (Fig. 4) have distinguished infarcted and normal
myocardial nuclei; we found that apoptotic cardiomyocytes were mainly distributed
in the peri-infarct region of myocardial microinfarction. The apoptosis rate was
dramatically higher in the CME group than in the Sham and CME + HUK groups (both
p
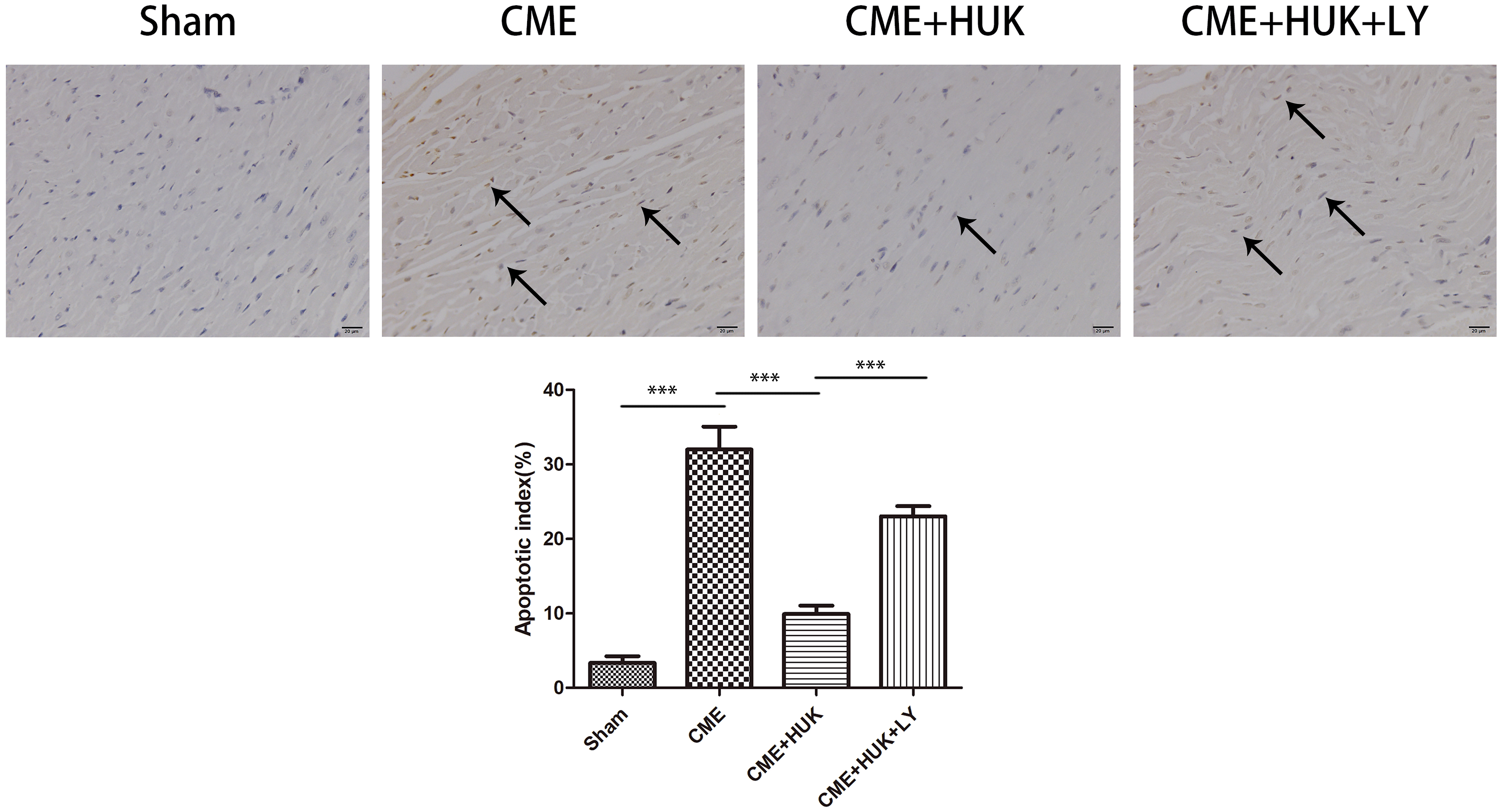 Fig. 4.
Fig. 4.TUNEL staining results of the infarcted and normal cardiomyocyte
nuclei and the apoptosis rate of cardiomyocytes in each group (n = 6, per group).
The arrows indicated the microspheres. The nuclei of normal cardiomyocytes were
stained light blue, while apoptotic cardiomyocytes’ nuclei were stained
yellow-brown. Data were presented as the mean
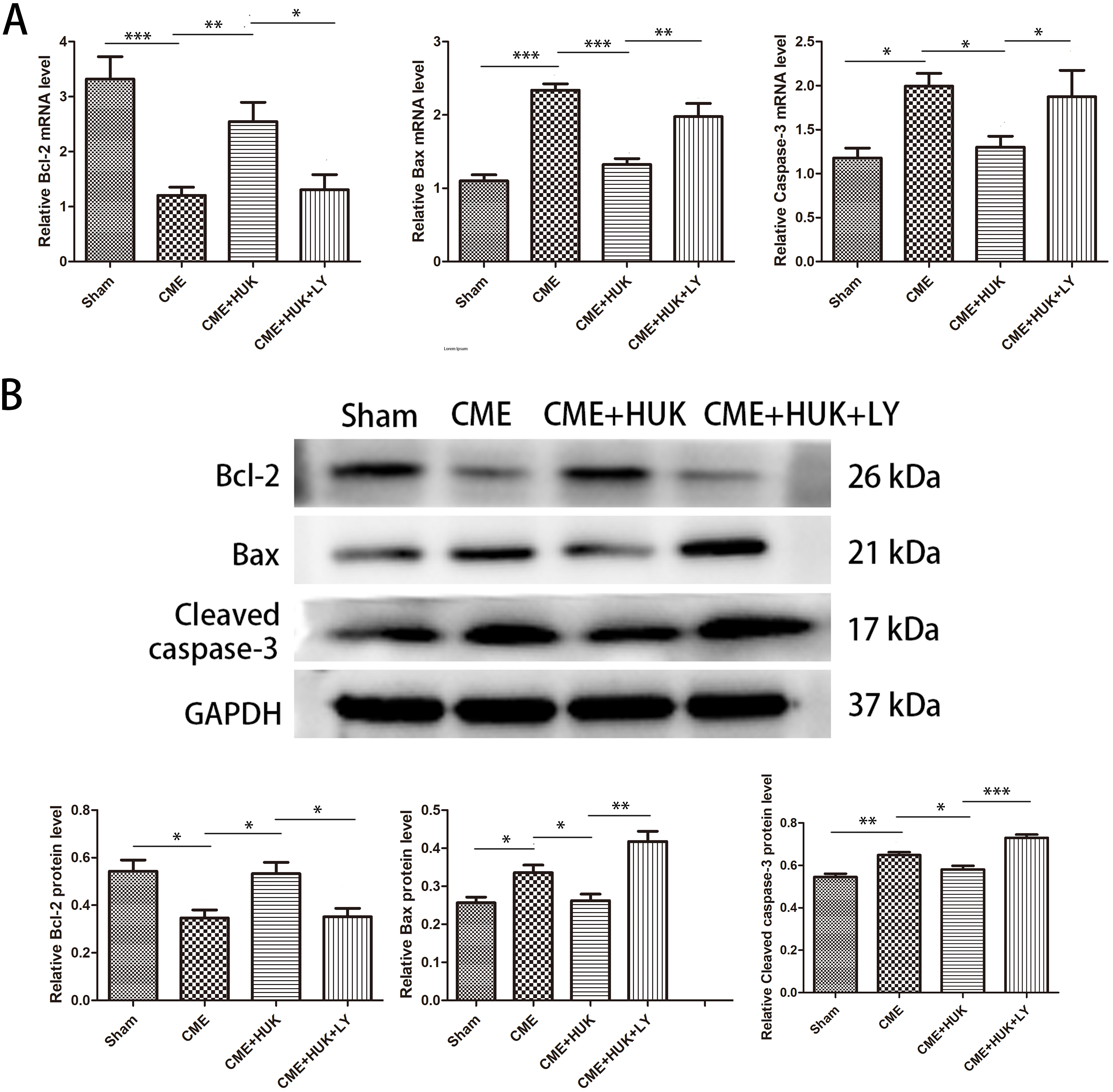 Fig. 5.
Fig. 5.Pretreatment with HUK attenuated CME-induced myocardial
apoptosis. (A) The mRNA expression levels of Bcl-2, Bax, and caspase-3 in cardiac
tissues (n = 6, per group). (B) The protein expression levels of Bcl-2, Bax, and
cleaved caspase-3 in cardiac tissues (n = 3, per group). Data were presented as
the mean
Furthermore, we observed from the IF results that Bax and cleaved caspase-3 expression levels were significantly decreased in the Sham and CME + HUK groups compared to those in the CME and CME + HUK + LY groups, and Bcl-2 in the CME and CME + HUK + LY groups were significantly downregulated (Fig. 6).
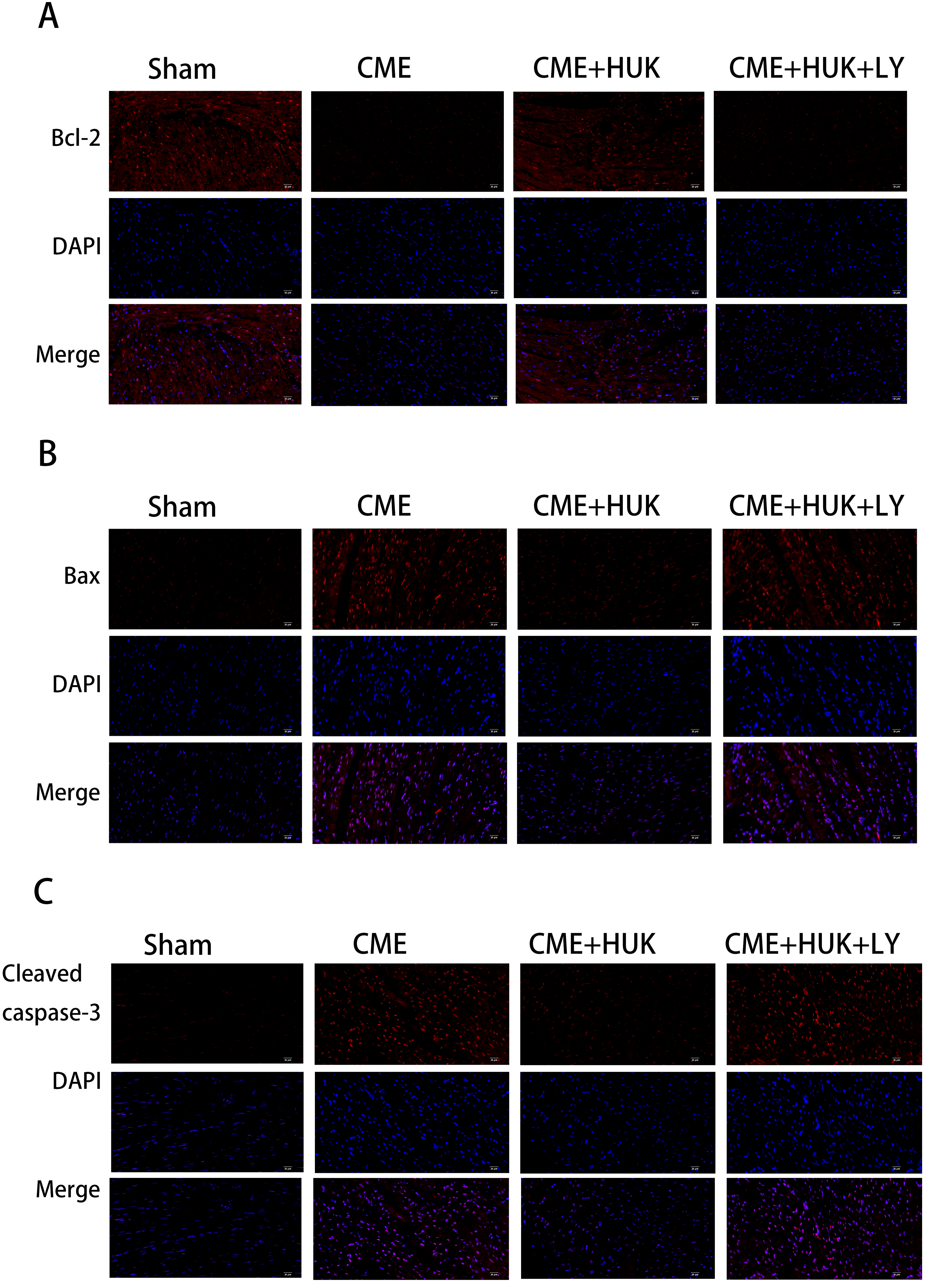 Fig. 6.
Fig. 6.Immunofluorescence staining of Bax, Bcl-2, and cleaved caspase-3
in cardiac tissues (magnification
To explore the effect of HUK on cardiomyocyte inflammation, two methods (ELISA
and western blotting) were used to identify the relative expression levels of
IL-1
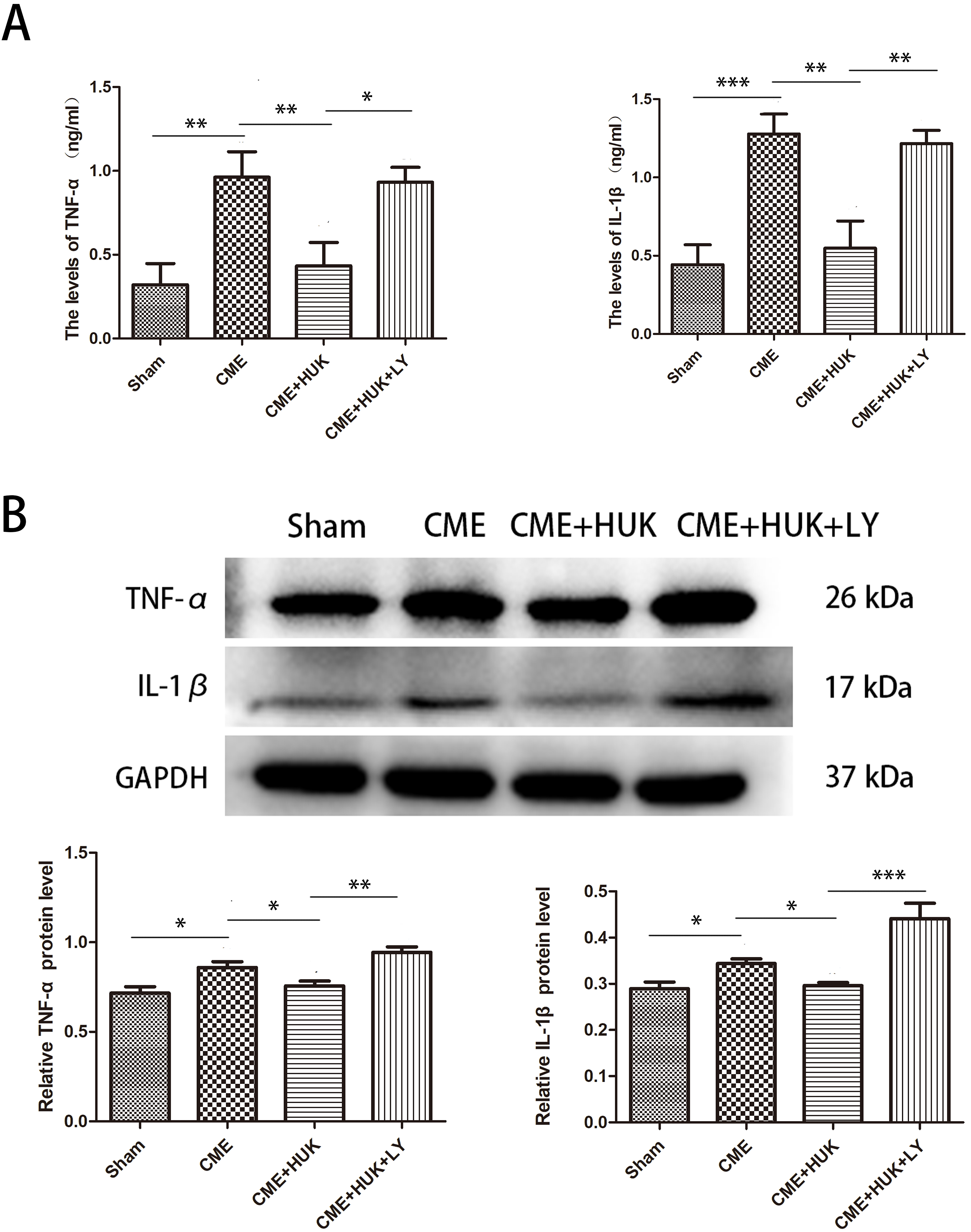 Fig. 7.
Fig. 7.Human Urinary Kallidinogenase reduced the myocardial
inflammation. (A) The serum levels of TNF-
In this study, we quantified serum myocardial injury biomarkers (including cTnI,
LDH, and CK-MB) between groups (Fig. 8A–C). Briefly, the degree of myocardial
damage was assessed by calculating the serum cTnI, LDH, and CK-MB levels. The CME
group had significantly higher serum levels of CK-MB, cTnI, and LDH compared to
the Sham group. In addition, serum LDH, cTnI, and CK-MB levels in the CME + HUK
group were significantly lower than those in the CME group. Compared with the CME
+ HUK group, serum CK-MB, cTnI, and LDH levels were significantly higher in the
CME + HUK + LY group (all p
 Fig. 8.
Fig. 8.Human Urinary Kallidinogenase reduced the serum levels of
myocardial injury markers (n = 6 per group). (A) The blood levels of cTnI in the
four groups. (B) The blood levels of LDH in the four groups. (C) The blood levels
of CK-MB in the four groups. Data were presented as the mean
Here, we determined the relative expression levels of PI3K/Akt/FoxO1 signaling
pathway marker genes (p-PI3K, PI3K, p-Akt, Akt, p-FoxO1, and FoxO1) at the
protein levels‘1 using western blotting (Fig. 9). This indicated that the
expression levels of p-PI3K, p-Akt, and p-FoxO1 in the CME group were
significantly lower than those in the Sham and CME + HUK groups (all p
 Fig. 9.
Fig. 9.Effect of HUK on PI3K/Akt/FoxO1-associated protein expression in
cardiac tissues (n = 3, per group). Data were presented as the mean
CME is a major cause of “no recurrent flow” in occluded coronary arteries after interventional procedures. The incidence of CME during perioperative PCI is 15%–20%, and it is as high as 45% in high-risk patients. CME may cause postoperative “no flow” or “slow flow” and microcirculation dysfunction, which in turn may lead to myocardial ischemia, arrhythmia, heart failure, and other consequences [16]. Given its high incidence, it has been viewed as an independent predictor of heart disease and mortality [17]. The aim of this study was to investigate the underlying mechanisms of HUK on CME. Our findings revealed that HUK activation of the PI3K/Akt/FoxO1 axis caused myocardial inflammation inhibition and anti-apoptosis, with the endpoint of ameliorating CME-induced myocardial injury.
HUK is an extract from urine, which was designed to block the post-stroke
inflammatory cascade and became a class I new drug in the USA [14].
Accumulating studies have shown that HUK was negatively associated with
TGF-
The PI3K/Akt signaling pathway is associated with the regulation of physiological functions such as apoptosis and inflammation. Its activation can reduce myocardial apoptosis and inflammation, thereby effectively reducing myocardial damage [20, 21, 22]. PI3K/Akt signaling pathway has mediated various biomolecules, including rapamycin (mTOR), glycogen synthase kinase 3 (GSK-3), forkhead box protein O1/3 (FoxO1/3), and nitric oxide synthase (NOS) [23]. He et al. [12] concluded that lipopolysaccharide located in the PI3K/Akt/FoxO1 pathway were beneficial to neuronal cell development, including injury mitigation, inhibition of apoptosis, and protection [24]. HUK was found to reduce apoptosis and inflammation in acute ischemic stroke by activating the PI3K/AKT/FoxO1 axis, thereby playing a protective role against brain injury. However, the effects of HUK on the PI3K/Akt/FoxO1 axis in CME and its role in CME have not been thoroughly studied.
Our previous studies showed that CME usually leads to cardiomyocyte apoptosis
and inflammation, which may cause cardiac dysfunction [6, 25]. For
cardiomyocyte apoptosis, the previous studies demonstrated substantial
cardiomyocyte apoptosis and abnormal expression of apoptosis-related genes
(up-regulated Bax and Cleaved caspase-3; down-regulated Bcl-2) in the infarct
zone [26, 27]. For cardiomyocyte inflammation, the expression levels of
inflammation-related genes (IL-1
We found that CME caused inflammation and increased apoptosis in rat
cardiomyocytes, leading to severe myocardial damage. HUK (pretreatment starting 7
days before CME) was involved in activating the PI3K/Akt/FoxO1 axis and
decreasing myocardial inflammation and apoptosis, thereby exerting a
cardioprotective effect in CME (shown in Fig. 10). In contrast, when treated CME
with the PI3K specific inhibitor LY294002, the essential proteins p-PI3K, p-Akt,
and p-FoxO1 were significantly downregulated, indicating that the PI3K/Akt/FoxO1
signaling pathway was suppressed. The expression levels of inflammation-related
genes (IL-1
 Fig. 10.
Fig. 10.Mechanism chart of HUK exerting cardioprotective effect in CME through PI3K/Akt/FoxO1 axis, created with BioRender (https://biorender.com/). Abbreviations: CME, coronary microembolization; HUK, Human Urinary Kallidinogenase.
This study had a few limitations. Firstly, the plastic microspheres of the CME rat model were not equipped with biological characteristics for atherosclerotic plaques (thrombosis, vasoactivity), and could not be equated with clinicopathological changes. Secondly, we investigated the effect of HUK on the inhibition of myocardial inflammation and apoptosis induced by CME through the PI3K/Akt/FoxO1 signalling pathway. However, we only used the PI3K specific inhibitor (LY294002) to affect the PI3K/Akt/FoxO1 signalling pathway. PI3K downstream molecules must also be targeted to exclude specificity further and avoid serendipity. Finally, other signalling pathways may be involved; therefore, further in vitro, or in vitro studies are required.
In summary, the findings of this study identified that HUK antagonizes CME-induced myocardial injury and exerts substantial protection. These cardioprotective effects were mediated by the activation of the PIK/Akt/FoxO1 axis, which significantly inhibited myocardial inflammation and apoptosis. Therefore, we speculated that HUK could potentially be used clinically in the “golden time” of CME and pretreatment. This research has the potential to provide a certain reference value for novel ideas in HUK, which would guide CME-related clinical strategies.
The data used to support the findings of this study are available from the corresponding author upon request.
LL and JX conceived the project; JX and BHM designed the study; TL and BHM directed the study; YQX, JX, BLW drafted the manuscript; YHL, GQL and QQN was responsible for all data analysis; LL, JX revised the manuscript; All authors have read and agreed to the published version of the manuscript.
Animal experimental protocols and steps in our study according to the NIH Guidelines for the Use of Laboratory Animals and were approved by the Animal Ethics Committee of Guangxi Medical University (License number: 202101006).
This work was supported by the First Affiliated Hospital of Guangxi Medical University.
This work was supported by Innovative Research Team Projec of Guangxi Natural Science Foundation (Grant No. 2018GXNSFGA281006) & the National Natural Science Foundation of China (Grant No. 82170349).
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.