
†These authors contributed equally.
Academic Editor: Taeg Kyu Kwon
Background: Endoplasmic reticulum stress (ERS) is a key part
of the apoptotic cascade that is initiated after cerebral ischemia-reperfusion
injury and is very important for research on poststroke rehabilitation. In
addition, the unfolded protein response (UPR) plays an important role in ERS
because it activates downstream apoptotic signal transduction and induces
apoptosis through the glucose-regulated protein 78 (GRP78)/protein kinase R
(PKR)-like ER kinase (PERK)/activating transcription factor 4 (ATF4) pathway. The
Gua Lou Gui Zhi Decoction (GLGZD) ameliorated neuronal apoptosis of
ischemia-reperfusion injury caused by middle cerebral artery occlusion (MCAO) had
been proved in our previous study. The present study aims to underly the
regulatory ability of GLGZD in ERS-induced apoptosis mediated by the
GRP78/PERK/ATF4 pathway. Methods: GLGZD was analyzed by HPLC. The
effects of GLGZD were obversed on MCAO-induced ischemic rats. The cerebral
infarct volume was detected by 2,3,5-Triphenyl-2H-Tetrazolium Chloride (TTC) Staining. Terminal Deoxynucleotidyl Transferase-Mediated dUTP-Biotin Nick End Labeling (TUNEL) were used to detect apoptosis.
Transmission Electron Microscopy (TEM), Ca
Stroke occurs abruptly, charactered with high morbidity, high mortality, and a high recurrence rate. There were ischemic stroke and hemorrhagic stroke on the basis of its pathogenesis. Ischemic stroke is more common, accounting for approximately 80% of all strokes, in which blood perfusion of the brain is altered, the dynamic equilibrium of ions is destroyed, and the metabolism of nerve cells is disrupted, ultimately inducing neuronal apoptosis and, in some cases, necrosis [1]. Approximately 70–80% of patients with stroke are plagued by limb motor dysfunction following stroke; among these defects, limb spasm has a high incidence rate and has become a major obstacle to patients’ daily life and recovery [2]. Recent studies have indicated that the limb spasticity cause by stroke was related the loss control of upper motor neurons after ischemia, and the dissimilatory process in the superior spinal cord disappears. The inhibitory effect of the central nervous system on limb motor function is weakened, resulting in increased muscle tension. Cell apoptosis is the key cause of neuronal loss after stroke [3]. Therefore, in rehabilitation of stroke, one of the important therapy approaches is inhibiting ischemic neuronal apoptosis [4].
A variety of apoptosis studies have focused on mitochondria, and two
intracellular apoptotic signaling pathways have been identified: the
mitochondrial pathway and the death receptor pathway. According to recent
studies, the mitochondria-associated endoplasmic reticulum (ER) couples with a
variety of organelles in terms of structure and function, conducted cell
apoptosis through various pathways [5]. Changes in the intracellular environment
is critical for the activation of ER. Both oxidative stress and Ca
According to Traditional Chinese medicine recording, Gua Lou Gui Zhi Decoction (GLGZD) has been used in treating stroke and its sequelae for thousands of years. GLGZD is a classic formula in traditional Chinese medical work. In modern clinical practice, GLGZD is commonly used because of its efficacy in the treatment of limb spasm after stroke [9, 10, 11]. Our previous study established a high-performance liquid chromatography (HPLC) method to determinate the representative compounds in GLGZD [12, 13]. The delegate chemical components of GLGZD have been qualitatively and quantitatively analyzed, and include citrulline, gallic acid, protocatechuic acid, paeoniflorin, glycyrrhizic acid, liquiritigenin, and isoliquiritigenin. These components are the foundation of GLGZD, they were reported had good effects in alleviating brain injury after cerebral ischemia-reperfusion. Based on these results, the treatment of GLGZD of ischemia-reperfusion injury mediated by the combined actions of these components. Furthermore, our previous works have suggested that GLGZD significantly inhibits neuronal apoptosis and exerts a neuroprotective effect in vivo and in vitro [13, 14]. However, researchers have not reported whether the molecular mechanism of GLGZD is related to the regulation of ERS. Fistly, a suitable model of stroke was established in this study, we observed the therapeutic effects of GLGZD on ischemia-injured rats using middle cerebral artery occlusion (MCAO), and then further investigated whether GLGZD inhibits ischemic neuronal apoptosis through the GRP78/PERK/ATF4 signaling pathway activated by ERS, so as to clarify the mechanism of GLGZD on alleviating limb spasticity after stroke. Our findings provided a theoretical support for the efficacy of GLGZD in neurological rehabilitation after ischemic stroke.
Trichosanthis Radix, Cinnamomi Ramulu, Paeoniae Radix Alba, Glycyrrhizae Radix et Rhizoma, Jujubae Fructus and Zingiberis Rhizoma Recens were purchased from Beijing Tong Ren Tang Chinese Medicine Co., Ltd (Beijing, China). The mentioned Chinese medicinal materials were identified by Professor Yang Chengzhi from Fujian University of Traditional Chinese Medicine.
Seventy-five male Sprague-Dawley (SD) rats, weighing between 210 and 230 g, were
purchased from purchased from Shanghai SLAC Laboratory Animal Co., Ltd. (license,
SCXK (Hu) 2017–0005; Shanghai, China) and housed in the pathogen-free facility
in the SPF animal experiment center of Fujian University of Traditional Chinese
Medicine. Rats were maintained in a temperature-controlled facility at 22
Extraction of GLGZD was conducted according to the original composition recorded
in the “Jin Kui Yao Lu”. GLGZD was made up with 30 g of Trichosanthis Radix, 9
g of Cinnamomi Ramulu, 9 g of Paeoniae Radix Alba, 6 g of Zingiberis Rhizoma
Recens, 9 g of Jujubae Fructus, and 6 g of Glycyrrhizae Radix et Rhizoma. These
medicinal materials were crushed, sequentially extracted with 10 and 8 volumes of
ddH
 Fig. 1.
Fig. 1.Fingerprint of the GLGZD. 1, gallic acid; 2, protocatechuic acid; 3, albiflorin; 4, paeoniflorin; 5, glycyrrhizin; 6, liquiritigenin; 7, cinnamic acid; 8, cinnamaldehyde; and 9, glycyrrhizic acid.
The model of MCAO was conducted by an intraluminal block, as we usually do [13]. Firstly, anesthetized the rats by intraperitoneal injection of 2% sodium pentobarbital (60 mg/kg), and then inserted a 3–0 silicon rubber-coated nylon monofilamen into the internal carotid artery (ICA) (about 18 mm from the CCA bifurcation). The filament was partially removed (~1.5 cm) for reperfusion after embolization for 2 h. After modeling, all rats were transferred to a clean cage and kept warm. When rats were awake, neurological function was scored using the Zea Longa scale [15], and muscle tension was determined using the modified Ashworth method [16]. The scoring processes were performed by two experimenters who were familiar with the scoring criteria but not participated in the experiments, they completed scoring simultaneously and independently in a double-blind manner. A higher score indicates greater nerve injury severity.
Only rats with limb paralysis and neurological function scores of 2 to 4 were included in subsequent experiments. The total number male SD rats were seventy-five, of them twelve rats were randomly assigned for the sham operation, and the remaining rats were used as the operation group. Five rats in the operation group had no neurological deficits or muscular hypertonia and were subsequently excluded from the experiment, and nine rats died during the experimental procedures. The rest forty-nine rats were successfully modeled rats.
Rats that were successfully modeled assigned randomly into four groups after
MCAO modeling (n = 12). The rats in the MCAO group were administered saline. Rats
in the GLGZD low-dose group (GLGZD-L) were administered GLGZD at the
concentration of 0.36 g
The CatWalk test is a sensitive and reliable method to detect motor function deficits in rats. One week before the test, the rats in this study were trained to pass a Catwalk channel smoothly in 10 s in a dark room environment. The movement of the rat was evaluated using CatWalk gait analysis (XT10.0, Noldus Information Technology Co., Ltd., Wageningen, The Netherlands) 1 h after the last treatment was administered. During the measurement, footprint refraction technology with an internal light source was used to perform efficient computer processing of the footprints recorded by video, and hence, the gait parameters of each mouse were evaluated.
The brains were excised rapidly after rats were decapitate. The brain parenchyma (after discarding the olfactory bulb, cerebellum and lower brain stem) was precooled with a –20 °C refrigerator for 5 min, and cut into 2-mm slices using stainless steel brain matrices. The slices were incubated in a closed container with TTC solution (Sigma, St. Louis, MO, USA) at 37 °C for 30 min in the dark, during incubation flipped sections every 10 min for evenly dyed. Images of cerebral infarct were captured with a digital camera (Nikon, Tokyo, Japan). Infarct volume of each section was traced and calculated using Image-Pro Plus 6.0 software (Media Cybernetics, Rockville, MD, USA).
Rats were anesthetized after the last administration. The thorax was opened and
a perfusion needle was fixed on the left ventricle via the apex of the heart,
and a small opening in the right atrial appendage was cut for perfusion. Then,
mice were successively perfused with ice-cold saline and fixative containing 4%
paraformaldehyde (PFA)/PBS. Brain tissues were excised for another 24 h fixing,
and subsequently embedded. The paraffin-embedded brains were cut into 4
Rat cerebral tissue in a volume of 1 mm
Paraffin sections (4
Paraffin sections (4
The ischemic cerebral cortex was isolated from the excised brains. 0.25%
trypsin was added in two hundred micrograms of tissue for digestion under a 37
°C-water bath for 30 min. Three volumes of precooled PBS were added
to terminate digestion. Collected the cell suspension and filtered through a
300-mesh nylon filter. The filtrate was centrifuged at 500
The brain was quickly excised after rats were sacrificed, and the ischemic cortex was isolated. The cortex samples were ground to a powder with liquid nitrogen.
For total proteins extraction, every one hundred milligrams of the tissue powder were lysed by 1 mL of RIPA lysis buffer (Beyotime Biotechnology, Shanghai, China) on ice bath for 30 min. Lysates were centrifuged at 14,000 rpm for 30 min at 4 °C. Collected the supernatant to obtain total protein.
For nucleoproteins extraction, every One hundred milligrams of the tissue powder
were added in 1 mL of cytosolic extraction reagent (Beyotime Biotechnology,
Shanghai, China) and the sample was placed on ice for 20 min. Then centrifuged
the extraction of samples at 14,000
The proteins concentrations were obtained by the BCA method after extracting
protein from each sample lysate. Then the protein sample was added loading buffer
and boilled at 100 °C for 10 min-denaturation. Proteins (50
Data obtained were statistically analyzed with SPSS software, version 20.0
(SPSS, Inc., Chicago, IL, USA), and presented as the means
The chromatographic fingerprint was acquired by qualitative and quantitative HPLC analysis (Fig. 1). In the extraction of GLGZD, 9 main compounds including gallic acid, protocatechuic acid, albiflorin, paeoniflorin, glycyrrhizin, liquiritigenin, cinnamic acid, cinnamaldehyde and glycyrrhizic acid were detected.
Neurological deficits of the MCAO-injured rats were assessed using Longa’s
method, and muscle tension was scored with the modified Ashworth scale on days 1,
3, 5 and 7 after modeling. Compared with the sham group, there were significant
increases of the neurological function score, muscle tension score and cerebral
infarct volume in the MCAO group (p
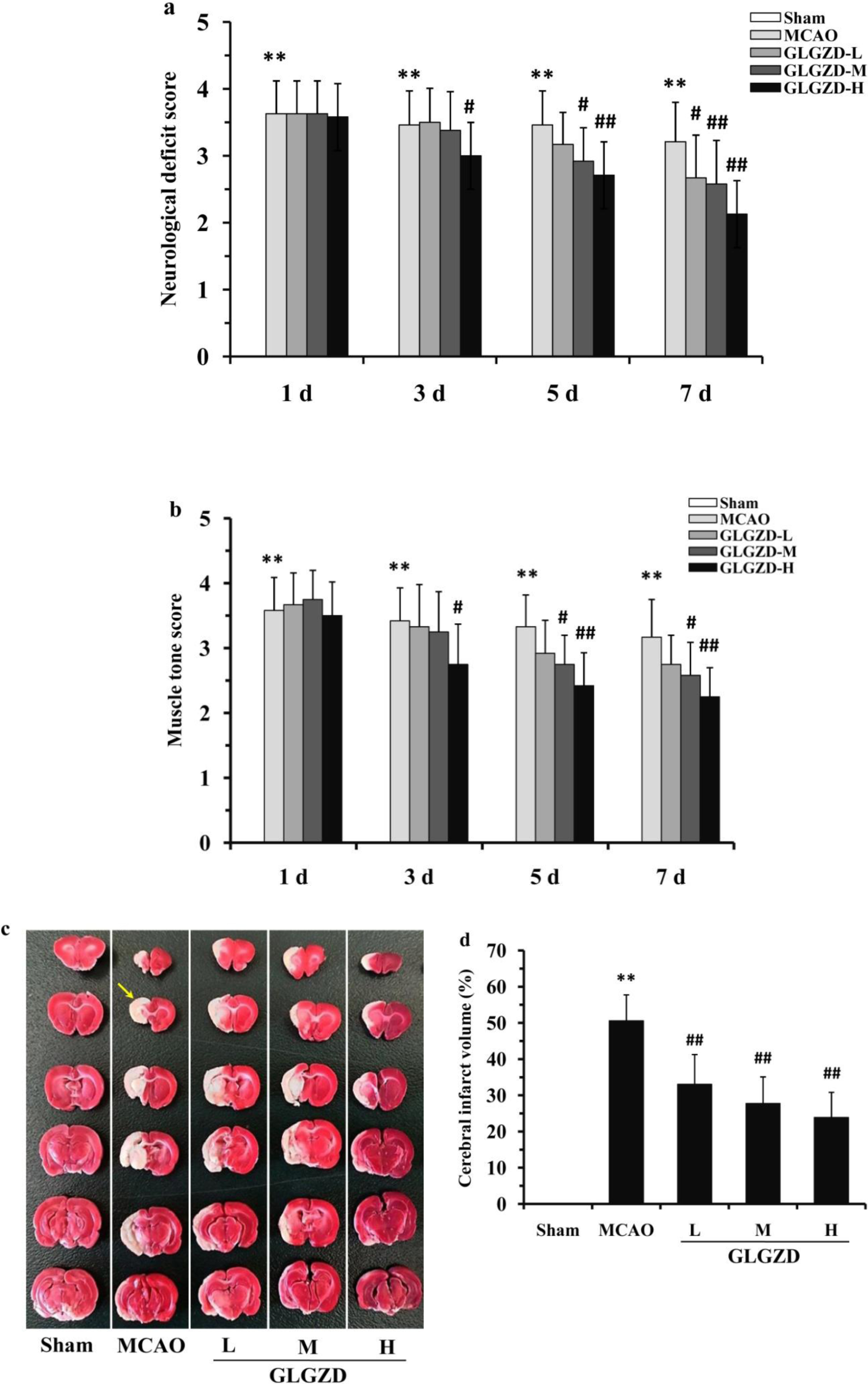 Fig. 2.
Fig. 2.GLGZD alleviates neurological deficits, muscle tension and the
cerebral infarct volume in MCAO-injured rats. (a) Neurological function scores.
(b) Muscle tension scores. (c) Representative images of TTC-stained cerebral
slices, yellow arrows indicated the “infarct zone”. (d) Quantitative analysis
of the cerebral infarct volume. All data are presented as the means
Results in Fig. 3 indicated that paralysis of the right front limbs of the MCAO
rats was obvious after cerebral ischemia reperfusion injury. It was found the
average running speed of MCAO rats was significantly decreased after surgical
operation (p
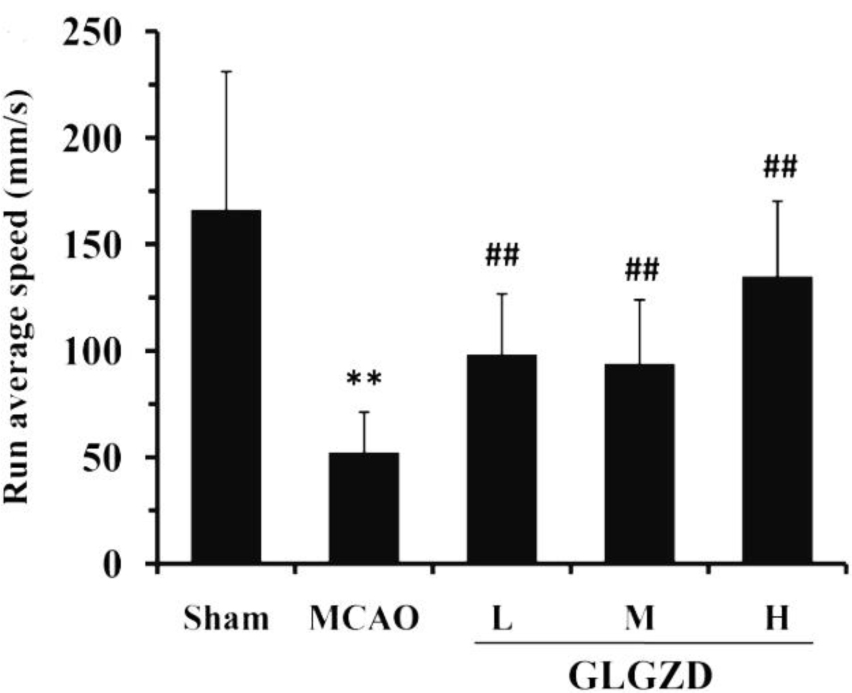 Fig. 3.
Fig. 3.GLGZD improves the motor function of rats after MCAO. The
CatWalk gait analysis system indicates the limb motor function of rats by
measuring the gait parameters. The CatWalk gait analysis system was used to
quantitatively assess the average running speed of the rats, indicating the limb
motor function of the rats. All data are presented as the means
Based on observations of TUNEL staining (Fig. 4a,b), compared with the sham
operation group, the TUNEL-positive cells in the cortex of MCAO rats were found
significantly increased (p
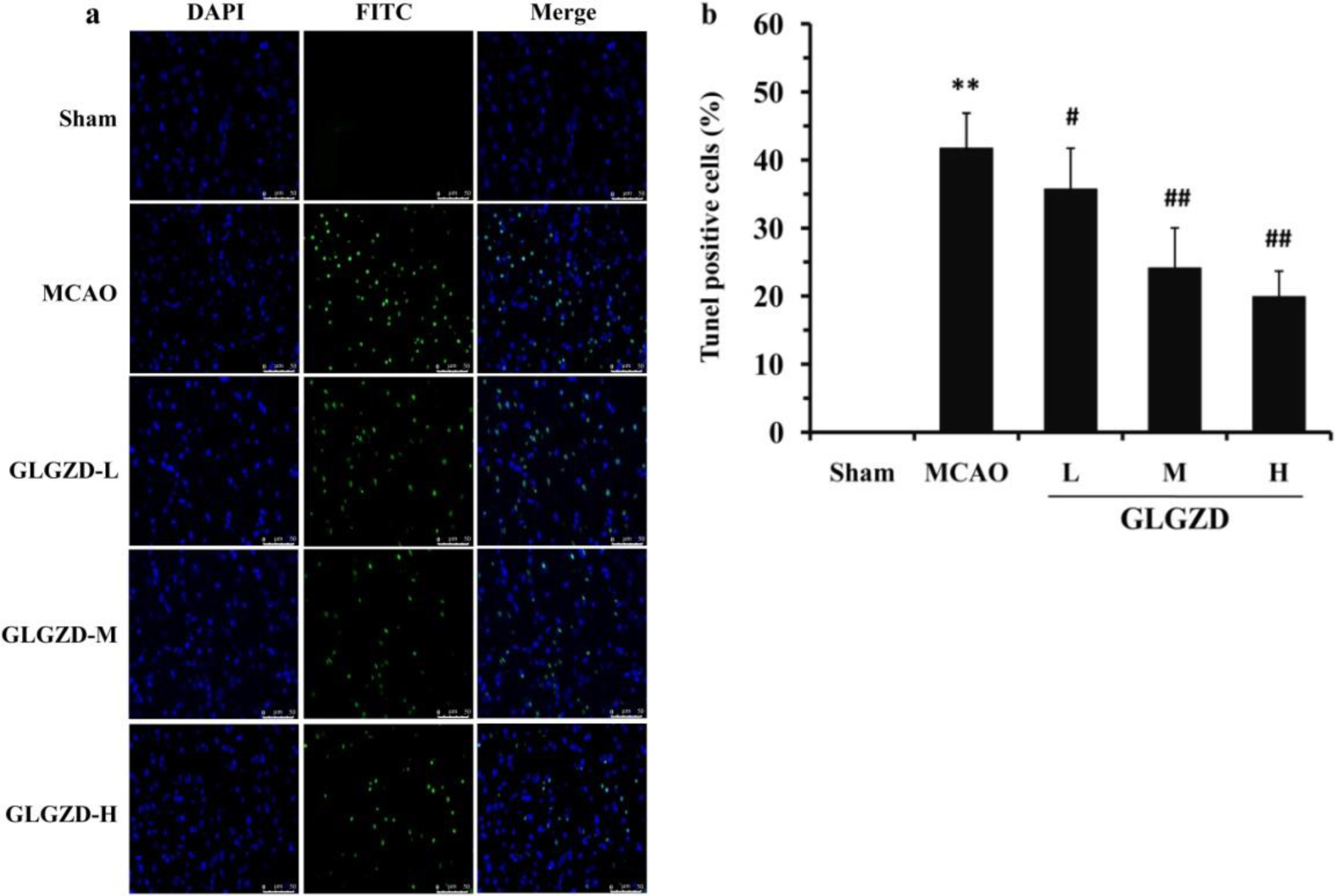 Fig. 4.
Fig. 4.GLGZD inhibits cell apoptosis in the ischemic cerebral cortex of
the rats after MCAO. The 3’-carboxy terminus is exposed when DNA is damaged, and
thus fragmented DNA in apoptotic cells exhibits green fluorescence after TUNEL
staining. The nuclei of the cells presented blue fluorescence after DAPI
staining. Only the cells labeled with both of these fluorescence stains were
considered positive cells. (a) Representative morphology of the apoptotic cells
(400
TEM images are displayed in Fig. 5. The nuclear membrane structure of the sham
group was clear and complete, rough ER was abundant. In addition, the cell
arrangement was neat and concentric. The ER of the MCAO group was significantly
decreased and disordered. The ER structure was altered, and expansion, swelling,
and vacuolization were apparent. However, the structure and function of the rough
ER were significantly ameliorated after GLGZD treatment, and the number of rough
ER was predominantly upregulated (p
 Fig. 5.
Fig. 5.ER morphology in the ischemic cerebral cortex of MCAO-injured
rats viewed using TEM; (10000
The results obtained with the fluorescence probe are shown in Fig. 6a,b, they
indicated that the Ca
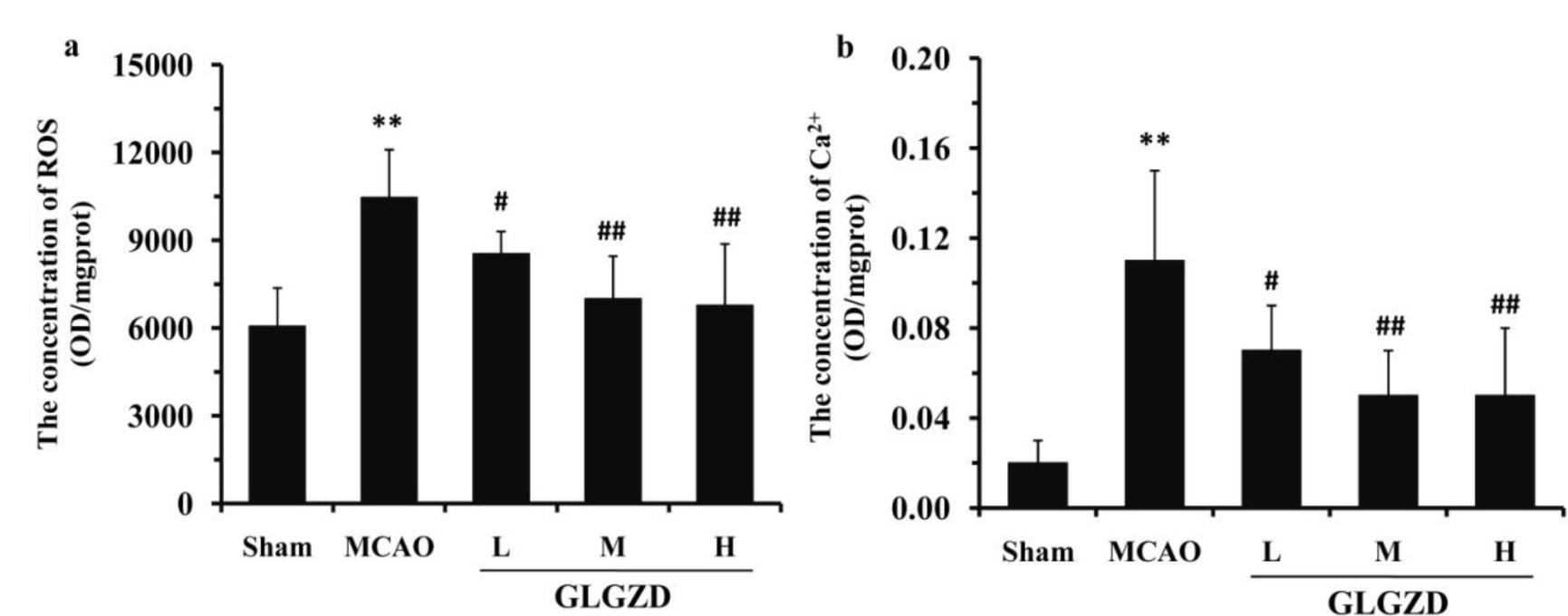 Fig. 6.
Fig. 6.Effects of GLGZD on the Ca
Immunohistochemistry staining (Fig. 7a,b) demonstrated that the IP3R, a
protein mainly located in the nucleus and cytoplasm, increased significantly in
the ischemic cortex of the MCAO group compared with the sham operation group
(p
 Fig. 7.
Fig. 7.Effects of GLGZD on the expression of the IP3R protein in the
ischemic area of the rats after MCAO. (a) The IP3R protein was stained using
immunohistochemical method, with the positively stained protein appearing brown.
The expression and location of IP3R were observed using electron microscopy
(400
Western blot analyse are shown in Fig. 8a–f, the levels of the total GRP78,
ATF4, CHOP, Ero-1
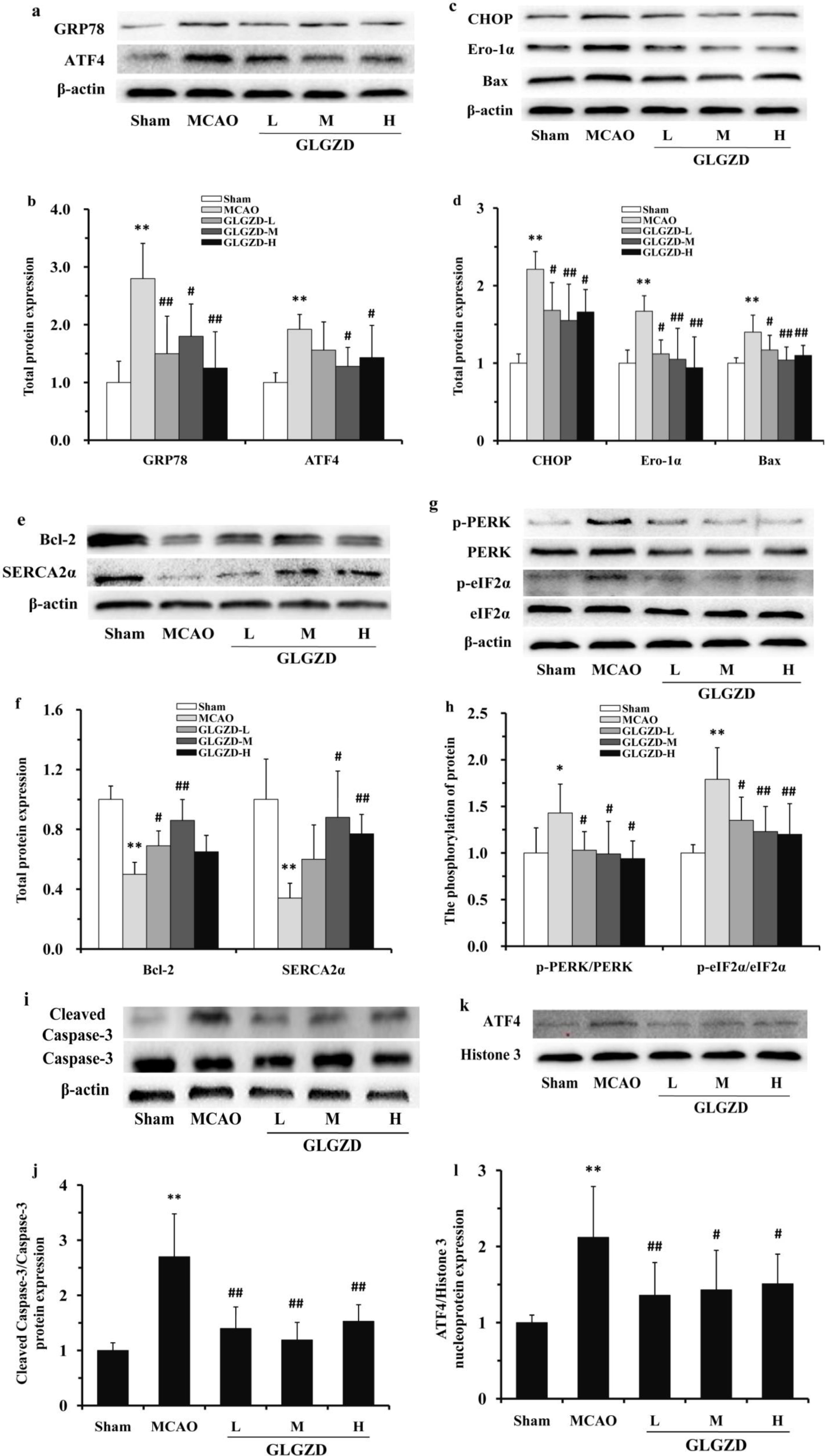 Fig. 8.
Fig. 8.Effects of GLGZD on the expression of proteins in the PERK/ATF4
signaling pathway-related the ischemic cortex of the rats after MCAO.
Representative Western blots (a), (c), (e) and the relative optical densities
(b), (d), (f) of the total protein levels of GRP78, ATF4, CHOP, Ero-1
As shown in Fig. 8g,h, compared the MCAO group with the sham group, a
significant increase of the phosphorylated form of
PERK and EIF2
As shown in Fig. 8i,j, compared the MCAO group with the sham group, the
level of cleaved caspase-3, a marker of apoptosis, was significantly increased in
the ischemic cortex of rat (p
As shown in Fig. 8k,l, compared the MCAO group with the sham group, the
nuclear expression of ATF4 was significantly increased in the ischemic cortex of
rat. After 7 days administration of GLGZD, the nucleoprotein ATF4 expressed level
in ischemic cortex was significantly lower than that in the MCAO group (p
Immunofluorescence staining (Fig. 9a,b) showed that the
GRP78 was mainly located in the cytoplasm. The ischemic cortex of the MCAO group
had a significantly higher expression of GRP78 than that of the sham group
(p
 Fig. 9.
Fig. 9.Effects of GLGZD on GRP78 protein expression of in the ischemic
cortex of rats after MCAO. (a) Immunofluorescence staining showing the location
and expression of the GRP78 protein (400
The blood supply zone in middle cerebral artery is a core region affected by ischemic cerebrovascular disease. Hence, A MCAO model was established, by inserting a standard microballoon catheter intraluminally from the common carotid artery into the middle cerebral artery proximal segment, to mimick clinical cerebral ischemic diseases [17]. Neurological deficits were assessed by calculating neurological function scores typically used as scoring standards in scientific analyses. The method we used is one of the most widely used methods for judging whether a MCAO model has been successfully established in experimental research [18]. Longa’s 5-point scoring system is recognized as one of the scoring standards of neurological function. The upper motor neuron injury after cerebral ischemia led to limb spasticity, which is a motor dysfunction after stroke. The spasticity is followed by excitability of motor circuits and an increase in muscle tension in limbs [4]. Therefore, muscle tension was assessed with the modified Ashworth scale, which was also used to indirectly indicate the limb spasticity of the rats.
In our experiment, when blocking the middle cerebral artery of the MCAO rats by
a thread plug, neurological deficits in the rats were observed. For example, rats
fell to the opposite side when walking, and some rats cannot walk without
assistance. The MCAO operation significantly increased the muscle tension score
and cerebral infarct volume. The muscle tension scores were all
Apoptosis was reported the main type of cell death caused by
ischemia-reperfusion injury in the ischemic penumbra cortex, it is the
culmination of a programmed cell death process controlled by genes. Apoptosis in
ischemia-reperfusion injury is intimately involved in the development
and expansion of cerebral infarction [19]. In our study, the neurons showed a
relatively complete cellular architecture and potential metabolic capability,
indicating that the apoptosis process in the MCAO model was reversible. According
to these characteristics, the infarct focus might be reduced and the neurological
function of patients with ischemic injury might be improved [20, 21]. Based on
results of TUNEL staining, we found a significant increase in TUNEL-positive
cells in the ischemic side of the rats’ cortex when compared the MCAO group with
the sham group (p
The ER is a subcellular organelle composed of a continuous membrane. The ER is a multifunctional organelle involved in various physiological activities. It is critical for maintaining intracellular homeostasis, especially in intracellular protein synthesis, calcium storage [22]. The ER is well-developed and abundant in neurons. However, the pathological changes in the ER after cerebral ischemia-reperfusion injury led to ER dysfunction, namely ERS, which leads to the dysfunction in synthesis, modification and folding of protein in the ER. The inactive proteins were largely accumulated in the ER lumen as a result. Therefore, the normal physiological activities of cells are affected, eventually leading to apoptosis [23]. Ultrastructural observations of the cells in the MCAO rats’ ischemic cortex using TEM showed that the number of rough ER decreased significantly. The structure altered, with expansion, swelling, and vacuolization appeared. After GLGZD administration for 7 days, the pathological state of the rough ER in the ischemic cerebral cortex was significantly ameliorated.
Previous researchers have investigated that oxidative stress and intracellular
Ca
The Ca
Our results obtained through staining with the fluorescence probe showed that
the Ca
The accumulation of unfolded proteins is a major manifestation of ERS. When
excessive external stimulation disrupts the functions of the ER, ERS induces
misfolding or unfolding of proteins. The acceleration of ERS and activation of
the UPR was following the proteins accumulated in the ER lumen. The UPR is both a
trigger of ERS and a key prosurvival process of ERS. The GRP78/PERK/ATF4
signaling is the main pathway regulating the protein balance and Ca
PERK is a type I transmembrane protein in the ER, which has an N-terminal region
with endonuclease activity, and this region detects and targets signals emitted
by the ERS. The C-terminus of PERK is located in the cytoplasm and has a
self-phosphorylation site. Under normal physiological conditions, the molecular
chaperone of GRP78 binds PERK in a complex, which is in a low activity state.
Once ERS occurs, the unfolded proteins accumulated in the ER lumen competed with
PERK in binding to GRP78. It results in the complex of GRP78 and PERK uncoupled.
This uncoupling causes GRP78 to be activated, and proteins are expressed in large
quantities [32]. Therefore, the upregulation of GRP78 expression is regarded as a
marker of ERS. In this research, both Western blotting and immunofluorescence
staining were used to determine the protein level of GRP78. It was observed
that the cytoplasm mainly expressed protein GRP78 was obviously more in the
ischemic cortex of the MCAO group than the sham group (p
PERK is the initiator protein of UPR, that is to say the UPR signal transduction
is mainly mediated by PERK. Under basal conditions, the active site of PERK is
sheltered by GRP78 and inhibited. Under ER stress conditions, degradation of the
GRP78-PERK complex releases PERK and exposes the dimerization site, which
promotes the auto-phosphorylation and dimerization of PERK, leading to the
downstream target protein eukaryotic translation initiation factor 2a (eIF2a)
aggregated and phosphorylated at serine 51 [33]. The affinity with the guanine
nucleotide exchange factor eIF2
Phosphorylation of eIF2
The mechanisms of CHOP mediate apoptosis are complicated. A study of an
in vitro ERS model showed that Ero-1
To sum up that GLGZD significantly decreased the cerebral infarct volume in
ischemic brain, ameliorated the pathological damage of cerebral
ischemia-reperfusion injury on cortical neurons, improved neurological and motor
functions after ischemia injury, and promoted the restoration of the quantity and
morphology of the rough ER in cells of the ischemic side of the cortex. GLGZD
reduced the ischemic cortex cells apoptosis, promoted the recovery of nerve and
alleviated limb spasticity after cerebral ischemia injury. The protection of
GLGZD turned out to be closely related to the modulation on the expression and
translocation of proteins in the GRP78/PERK/ATF4 pathway (Fig. 10). GLGZD acted
as an effective neuroprotective agent through the reduction of oxidative
stress-induced injury and the promotion in a dynamic balance of Ca
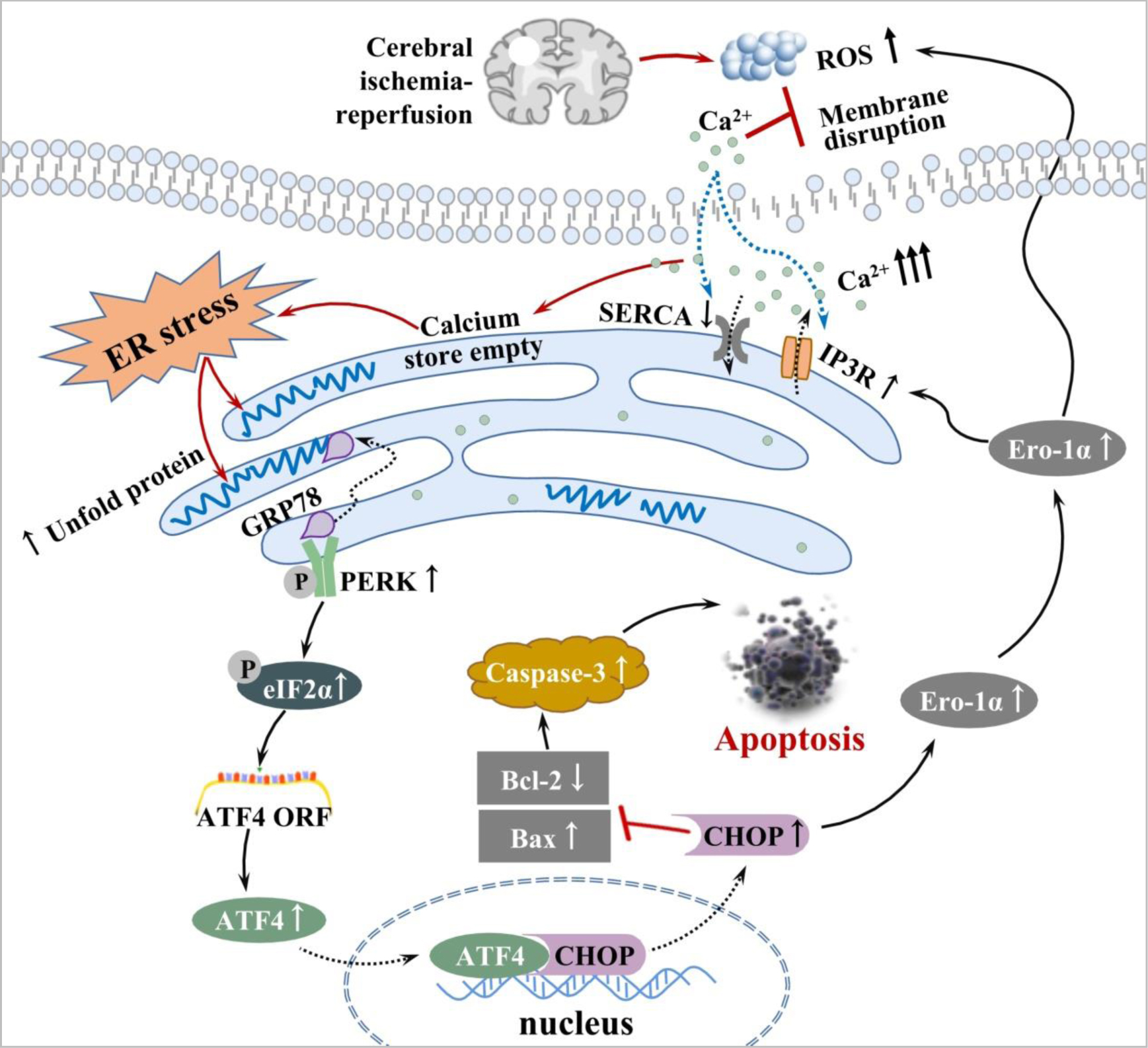 Fig. 10.
Fig. 10.The underlying mechanisms by which GLGZD attenuates apoptosis by inhibiting ERS after MCAO. Schematic diagram illustrating the involvement of the GRP78/PERK/ATF4 signaling pathway in the effects of GLGZD on ERS-induced apoptosis in the MCAO model.
MCAO, Middle cerebral artery occlusion; GLGZD, Gualou-Guizhi Decoction; GRP78,
Glucose-regulated protein 78; ER, Endoplasmic reticulum; ERS, Endoplasmic
reticulum stress; UPR, Unfolded protein response; PERK, Protein kinase R
(PKR)-like ER kinase; Ero-1
LN and MH supervised the work; YChen and ZC designed the experiments and wrote the manuscript; YCao and WC performed the experiments; WX and WL provided help and advice on conception; YZ analyzed the data and reviewed the manuscript. All authors discussed the results and commented on the manuscript. All authors provided critical comments on the manuscript. All authors read and approved the final manuscript.
This study was approved by the Ethics Committee of Fujian University of Traditional Chinese Medicine (Fuzhou, China), the ethic approval number is: FJTCM IACUC 2021086.
Not applicable.
This study was funded by the National Natural Science Foundation of China [grant number 81873031], the Science Foundation of the Fujian Province, China [grant number 2021J01926], and the School Fund of Fujian University of Traditional Chinese Medicine [grant number X 2019007].
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.