





Frontiers in Bioscience-Landmark (FBL) is published by IMR Press from Volume 26 Issue 5 (2021). Previous articles were published by another publisher on a subscription basis, and they are hosted by IMR Press on imrpress.com as a courtesy and upon agreement with Frontiers in Bioscience.

1 Department of Gastroenterology, Beijing Friendship Hospital, Capital Medical University, National Clinical Research Center for Digestive Disease, Beijing Digestive Disease Center, Beijing Key Laboratory for Precancerous Lesion of Digestive Disease, Beijing, 100050, P. R. China
Abstract
Asporin (ASPN), a member of small leucine-rich repeat proteoglycan (SLRP) family of proteins, serves important roles in diverse biological responses and disease conditions. We tested the hypothesis that ASPN regulated proliferation of gastric cancer (GC) cells and identified its down-stream regulators. ASPN promoted the proliferation of GC cells. We identified the effector of this effect as proteasome 26S subunit non-ATPase 2 (PSMD2) which is known to regulate proliferation through suppression of DUSP7, WIP1 and PTEN and then inducing the phosphorylation of ERK, P38 and AKT. PSMD2 co-immunoprecipitated with ASPN from GC cell lysates and co-localized with PSMD2 inside GC cells. Moreover, knockdown of ASPN significantly increased the expression of DUSP7, WIP1 and PTEN and led to a repression in the phosphorylation of ERK, P38 and AKT. These changes were counteracted by knockdown of PSMD2. In conclusion, ASPN promotes cell proliferation by interacting with PSMD2, and down-regulation of its effectors and serves as a potential therapeutic target in GC.
Keywords
- Asporin
- PSMD2
- Proteasome
- Proliferation
- Gastric cancer
Gastric cancer (GC) is one of the most common malignant carcinomas worldwide, with a high incidence and mortality rate, especially in East Asia (1, 2). Despite advances in molecular biological technology, the mechanism of GC progression remains unclear (3, 4). A few molecules have been identified as clinical treatment targets according to their vast oncogenic effects in GC (5, 6). However, GC is a disease with high heterogeneity, and molecular therapies targeting these molecules are still not sufficient for controlling the growth and progression of GC. Thus, exploring the molecular mechanism of GC tumorigenesis remains an important way of finding novel biomarkers and therapeutic targets.
The ASPN gene is localized on chromosome 9q22.31 and encodes a secretory protein containing 380 amino acids (7). ASPN belongs to the small leucine-rich proteoglycan (SLRP) family, which is the main component of the non-collagen extracellular matrix (ECM) (8, 9). In the physiological state, ASPN binds to collagen and induces collagen mineralization (10, 11). Several studies have recently reported that in the microenvironments of breast, prostate, pancreas, colorectum and scirrhous gastric cancers, ASPN secreted by cancer-associated fibroblasts (CAFs) or cancer cells enhanced the invasion and migration of cancers by regulating the epithelial to mesenchymal transition (EMT) or interacting with cell-membrane receptors (12-17). Thus, ASPN may play an important role in the tumor microenvironment of certain cancers, but the potential effects of ASPN found inside GC cells have not been illuminated. Ding et al. suggested that ASPN participated in GC cell growth and migration via regulating EGFR signaling and the apoptosis pathway (18). However, it is generally accepted that the mutation of RAS (an EGFR downstream effector) is partly present in various subtypes of GC. Moreover, the AGS cell line, which were used for the mechanism research in the above report, has been reported for containing the mutation of RAS many times (19, 20). Thus, novel signaling pathways still require further exploration to explain the mechanism by which ASPN regulates the phosphorylation of EGFR and ERK.
26S proteasome non-ATPase regulatory subunit 2 (PSMD2) was identified as an important non-ATPase subunit of the 19S regulator lid of the proteasome, which can recognize and bind to substrates (21). Protein ubiquitination and degradation by the proteasome play fundamental roles in the regulation of cell growth, cell cycle, apoptosis, differentiation, gene transcription and signal transduction (22, 23). As reported, the proteasome could promote cell proliferation by degrading some tumor suppressor factors, such as DUSP7, WIP1 and PTEN, which could repress the phosphorylation of ERK, P38 and AKT respectively (24-27). As a vital functional component of the proteasome, PSMD2 could also be a potential oncogenic effector and drug target in tumor development and progression. Yunhai et al. observed that PSMD2 regulated cell proliferation and the cell cycle by modulating P21 and P27 in breast cancer (28). In lung cancer, PSMD2 was found to be associated with poor prognosis and could promote cell growth via regulating the P38 and AKT pathways (29). Considering the fundamental effects on tumor progression, inhibition of the proteasome has been studied as a novel way to repress tumor progression (30, 31). Nevertheless, no study has revealed the function of PSMD2 in GC.
In this study, we reported that ASPN could promote proliferation and colony formation in GC cell lines. We also identified a strong interaction and colocalization between ASPN and PSMD2. Further studies showed that siASPN upregulated the expression of DUSP7, WIP1 and PTEN, and PSMD2 knockdown could mainly rescued the downregulation of ectopic ASPN on DUSP7, WIP1 and PTEN. Based on these findings, we suggested that ASPN could promote GC cell proliferation by interacting with PSMD2 and then promoting the degradation of downstream effectors.
The AGS cell line was acquired directly from American Type Culture Collection (ATCC), and SGC-7901 cells were purchased from Chinese Academy of Sciences (CAS). Dulbecco’s modified Eagle’s medium: nutrient mixture F-12 (DMEM/F-12) and Dulbecco’s modified Eagle’s medium (DMEM) were used as the culture medium for the two cell lines, respectively, supplemented with 10% fetal bovine serum (FBS) under standard culture conditions (5% CO2 at 37°C). Cell lines applied in the experiments were passaged less than 5 times and acquired within the past three years.
Lentiviruses with the full-length ORF of ASPN cloned into LV5 with the eukaryotic resistance of purine were purchased from Suzhou GenePharma company. Puromycin was added to the culture medium for incubation for at least 1 week to filter positive expression cells. Overexpression was identified via green light under a fluorescence microscope and western blot (WB) assays. ASPN and PSMD2 siRNAs were synthesized by GenePharma (Shanghai GenePharma Co., Ltd., Shanghai, China). A Lipofectamine 3000 kit (Life Technologies, USA) was applied for siRNA transfection assays according to the provided protocols. Scrambled RNA synthesized by GenePharma was used as a negative control, and the knockdown efficacy was also detected by WB. Sequences of siRNAs are listed in Table 1.
To evaluate the effects of ASPN on the cell viability of GC cells (AGS and SGC-7901), one-step MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) assays were performed. A total of 3000 cells of the two cell lines per well were seeded in four 96-well plates after overexpression or knockdown of ASPN. The samples were incubated with MTS for 2 h at time points of 6 h, 30 h, 54 h and 78 h after seeding and then tested using an enzyme-labeled meter (Spectramax M3, Molecular Devices).
For the colony formation assay, 1000 cells per well were plated in 6-well plates after siRNA transfection. After 10-14 days of incubation under standard culture conditions, visible colonies formed and were stained by crystal violet. The cells were washed with phosphate-buffered saline (PBS) until the excess dye cleared. The size and number of colonies were counted by ImageJ. Three independent experiments were performed in all assays.
According to the protocol of a Cell-Light™ EdU Apollo®567 Cell Tracking Kit (Ribobio, Guangzhou, China), 1.5 x 10^5 cells were seeded in 24-well plates after overexpression or knockdown of ASPN. Then, 50 μM EdU labeling medium (Reagent A) was added to the cell culture of the two cell lines (AGS and SGC-7901) and incubated under standard conditions for 4 h. Afterwards, cells were fixed with 4% paraformaldehyde and stained with 1X Apollo dyeing reaction solution and 1X Hoechst 33342 dyeing solution. The percentage of EdU-positive cells was observed under a fluorescence microscope, and cells were counted in nine random fields of at least three wells.
To detect the cellular localization of ASPN and PSMD2, AGS and SGC-7901 cells were plated on sterile coverslips in 6-well plates for at least 8 h. After washing with PBS 3 times, the cells were fixed in 4% paraformaldehyde for 10 minutes. Then, the cells were permeabilized with 0.3% Triton X-100 and blocked in 5% bovine serum albumin (BSA) for 1 h. Rabbit anti-ASPN and mouse anti-PSMD2 antibodies were mixed with PBS at a concentration of 1:50. Then, the mixed antibodies were added to the coverslips, and the cells were incubated overnight at 4°C. After incubation at room temperature with two types of fluorescent secondary antibodies (Alexa Fluor 594–conjugated anti-rabbit IgG and Alexa Fluor 488–conjugated anti-mouse IgG) (Life Technologies, USA) in the dark for 2 h, DAPI was used to stain the nuclei, and the cells were observed via confocal microscopy (IX83, FLUOVIEW FV1200, Olympus).
Specific lysis buffer for immunoprecipitation (IP) (HEPES 50 µM, NaCl 150 µM, EDTA 1 mM, 1% Triton, 10% glycerol) was used for the endogenous protein extraction of AGS cells. Cells were centrifuged at 1,2000 rpm for 30 minutes, and protein A/G agarose (Beyotime Biotechnology, China) was cleaned with the same lysis buffer 3 times. A total of 20 µl of resuspended agarose, 10 µg of primary antibody (anti-ASPN and anti-PSMD2) and 1 ml of extracted protein were mixed in a new EP tube and incubated on a shaker at 4°C overnight. Following centrifugation at 1000 rpm for 3 minutes and washing with lysis buffer at least 3 times, the remaining beads were added to 30 µl loading buffer and then denatured at 99°C for 10 minutes. To verify the interaction between ASPN and PSMD2, the complexes after denaturing were tested by Western blotting assays using anti-ASPN and anti-PSMD2 antibodies.
A bicinchoninic acid (BCA) protein assay kit (Thermo Fisher, USA) was applied to detect protein concentration. A total of 50 µg/30 µl denatured proteins per line were separated via electrophoresis. Then, the separated proteins were transferred to polyvinylidene fluoride (PVDF) membranes, which were blocked in 5% (w/v) nonfat milk for at least 2 h. Membranes were incubated on a rotator in diluted primary antibodies (Table 2) against ASPN, PSMD2, GAPDH, DUSP7, WIP1, PTEN, P38, p-P38, AKT, p-AKT, ERK, and p-ERK at 4°C overnight. After the primary antibodies were recycled the next day, the membranes were incubated in secondary antibodies for at least 1 h at room temperature. The enhanced chemiluminescence system (BIO-RAD, USA) and hypersensitive chemiluminescence reagents were used to detect the protein bands.
| Primary Antibody | Company | Catalogue number | Host | Clone | Dilution Factor | ||
|---|---|---|---|---|---|---|---|
| Western blotting | Immunoprecipitation | Immunofluorescence | |||||
| GAPDH | Proteintech | 60004-1-1g | Mouse | Monoclonal | 1:1000 | ||
| ASPN | Abcam | ab58741 | Rabbit | Polyclonal | 1:200 | 1:50 | 1:50 |
| PSMD2 | Santa Cruz | sc-271584 | Mouse | Monoclonal | 1:1000 | 1:50 | 1:50 |
| ERK | CST | 4695 | Rabbit | Monoclonal | 1:1000 | ||
| p-ERK | CST | 4370 | Rabbit | Monoclonal | 1:1000 | ||
| P38 | Proteintech | 14064-1-AP | Rabbit | Polyclonal | 1:1000 | ||
| p-P38 | CST | 4511 | Rabbit | Monoclonal | 1:1000 | ||
| AKT | Proteintech | 10176-2-AP | Rabbit | Polyclonal | 1:1000 | ||
| p-AKT | CST | 4060 | Rabbit | Monoclonal | 1:1000 | ||
| DUSP7 | Proteintech | 26910-1-AP | Rabbit | Polyclonal | 1:1000 | ||
| WIP1 | Proteintech | 26532-1-AP | Rabbit | Polyclonal | 1:1000 | ||
| PTEN | CST | 9188 | Rabbit | Monoclonal | 1:1000 | ||
All statistical analyses were conducted using GraphPad Prism5. Data represent means ± SD (standard deviation). T tests were conducted to evaluate the differences between two groups. P <0.05 was considered to indicate a statistically significant difference.
First, we designed WB assays to detect the knockdown effects of the two parallel siRNA sequences of ASPN in the two cell lines (AGS and SGC-7901). The results suggested that both sequences markedly reduced ASPN expression. In addition, siASPN2 showed better knockdown efficiency than siASPN1 (Figure 1A). Thus, we performed subsequent experiments with the sequences of siASPN2. To evaluate the effect of ASPN in GC cells, we performed colony formation and MTS assays in the two GC cell lines by ASPN knockdown. Colony formation assays and statistical analysis confirmed that ASPN increased the number and size of colonies in the two types of GC cells (AGS and SGC-7901) (Figure 1B). The growth curves of the two GC cell lines suggested that cell viability was significantly repressed by knockdown of ASPN (Figure 1C). EdU assays were conducted to evaluate the effect of ASPN on proliferation in both GC cell lines and revealed that knockdown of ASPN markedly reduced the number of EdU-positive cells (Figure 1D). To further support the conclusion that ASPN could promote cell proliferation in contrast to the possibility that ASPN is an inhibitor of proliferation, we performed a cell growth curve analysis and EdU assays by ectopic ASPN overexpression in AGS and SGC-7901 cell lines. The results verified that overexpression of ASPN promoted cell viability and proliferation (Figure 1E and F).
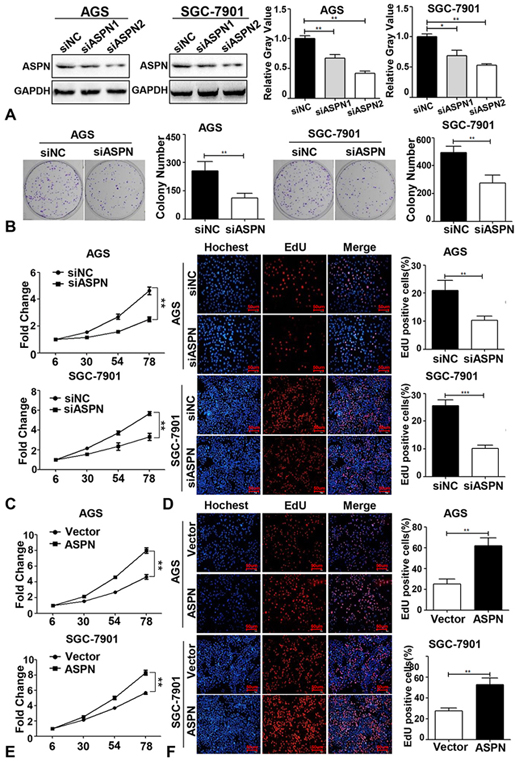 Figure 1.
Figure 1.ASPN promotes GC cell proliferation. (A) WB and the relative gray value (of control) analysis of the knockdown effects of two siASPN sequences (siASPN1 and siASPN2) in AGS and SGC-7901. (B) Knockdown of ASPN inhibited the growth of AGS and SGC-7901 cells (one step MTS growth curves were tested in 6, 30, 54, 78hours; Y-axis: cell viability fold change). (C) EdU assays showed that cell proliferation was repressed via knockdown of ASPN in AGS and SGC-7901 cells (DAPI: Blue; EdU: Red). (D) Ectopic ASPN promoted the cell viability of AGS and SGC-7901. (E) Overexpression of ASPN enhanced the ratio of EdU-positive cells in AGS and SGC-7901. (F) Knockdown of ASPN inhibited AGS and SGC-7901 cells colony formation (left panel: representative colony formation staining; right panel: quantitative analysis of colony number in three independent assays). All assays were performed in triplicate, and one representative result is displayed. Values are presented as the mean±SD. **P<0.01; ***P<0.001.
To explore the mechanism by which ASPN promotes proliferation in GC cells, we performed IP assays of SGC-7901 cells with ectopic ASPN. Successful purification of ectopic ASPN in SGC-7901 cells was demonstrated by the strong signal in the WB data (Figure 2A). Then, SDS polyacrylamide gels containing purified ASPN and its potential interacting proteins were separated by electrophoresis according to their different molecular weights and subjected to mass spectrometric analysis. The results showed that PSMD2 was identified as a potential binding protein of ASPN. Thus, we performed mutual co-IP assays to test the interaction between endogenous ASPN and PSMD2 in SGC-7901 cells under physiological conditions. Our results of the following WB analysis confirmed the strong interaction between ASPN and PSMD2 in GC cells (Figure 2BC). To further verify their interaction and location in GC cells, we detected the localization of ASPN and PSMD2 in AGS and SGC-7901 cells via immunofluorescence (IF) assays. As shown in Figure 2D, the colocalization of ASPN and PSMD2 inside GC cells was notably observed in both the cytoplasm and nuclei of AGS and SGC-7901 cells (DAPI: nuclei; Red: ASPN; Green: PSMD2). Taken together, these findings suggest that PSMD2 is a crucial copartner of ASPN.
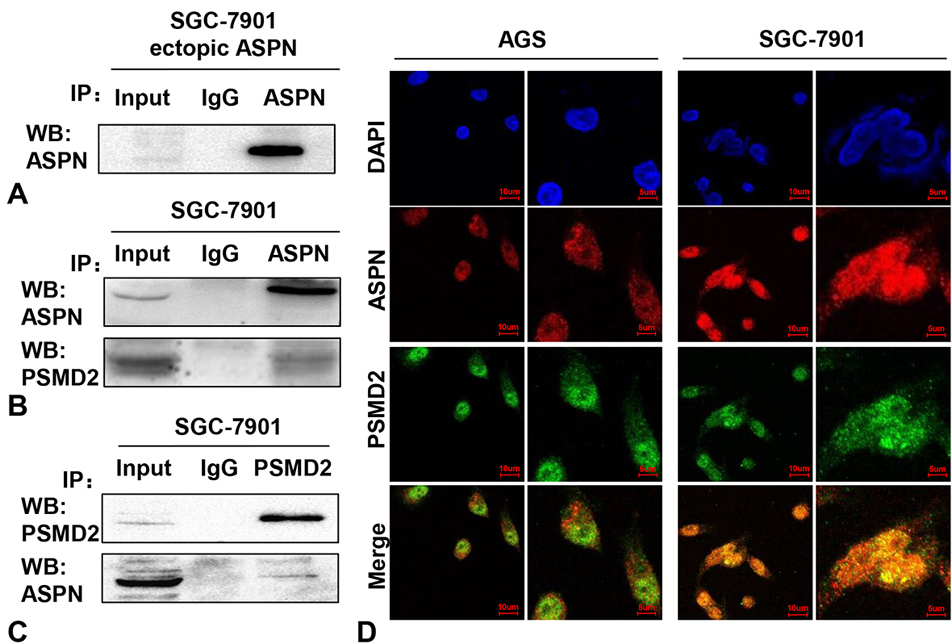 Figure 2.
Figure 2.ASPN interacts with PSMD2 in GC cells. (A) A markedly band was detected after ectopic ASPN being purified by anti-ASPN antibody via WB analysis. Co-IP assays were performed in SGC-7901 cells with both ASPN (B) and PSMD2 (C) antibodies, which consistently indicated a strong interaction between ASPN and PSMD2. Three samples were pipetted into three different lanes according to the antibodies added to or not added to the samples (Input group: no antibody; IgG group: IgG; PSMD2 group: anti-PSMD2 antibody; ASPN group: anti-ASPN antibody). (D) Obvious merged points validated the colocalization of ASPN and PSMD2 in AGS and SGC-7901 cell nuclei and cytoplasm via immunofluorescence staining (blue: DAPI; green: PSMD2; red: ASPN).
As reported, the proteasome promoted cell proliferation by enhancing the degradation of some tumor suppressor factors, such as DUSP7, WIP1 and PTEN in ERK, P38 and AKT signaling, respectively (24-27). Considering that PSMD2 is a subunit of the 19S regulator lid in the proteasome, which could promote the binding and degradation effects of the proteasome (32), it is possible that ASPN might participate in degradation of the proteasome on its downstream effectors (such as DUSP7, WIP1 and PTEN) by interacting with PSMD2. Thus, we performed a WB analysis of DUSP7, WIP1, PTEN, p-ERK, p-P38 and p-AKT by knocking down the expression of ASPN in AGS and SGC-7901 cells to evaluate whether ASPN could regulate the level of these downstream proteins of PSMD2. As shown in Figure 3A-E, downregulation of ASPN expression induced increased DUSP7, WIP1 and PTEN expression and decreased p-ERK, p-P38 and p-AKT expression. However, knockdown of ASPN did not reduce the protein level of PSMD2 (Figure 3C). These results suggest that ASPN regulated downstream DUSP7, WIP1 and PTEN levels by interacting with PSMD2 and forming a possible steric hindrance effect at the proteasome (inhibiting the degradation of DUSP7, WIP1 and PTEN) rather than directly regulating the expression of PSMD2.
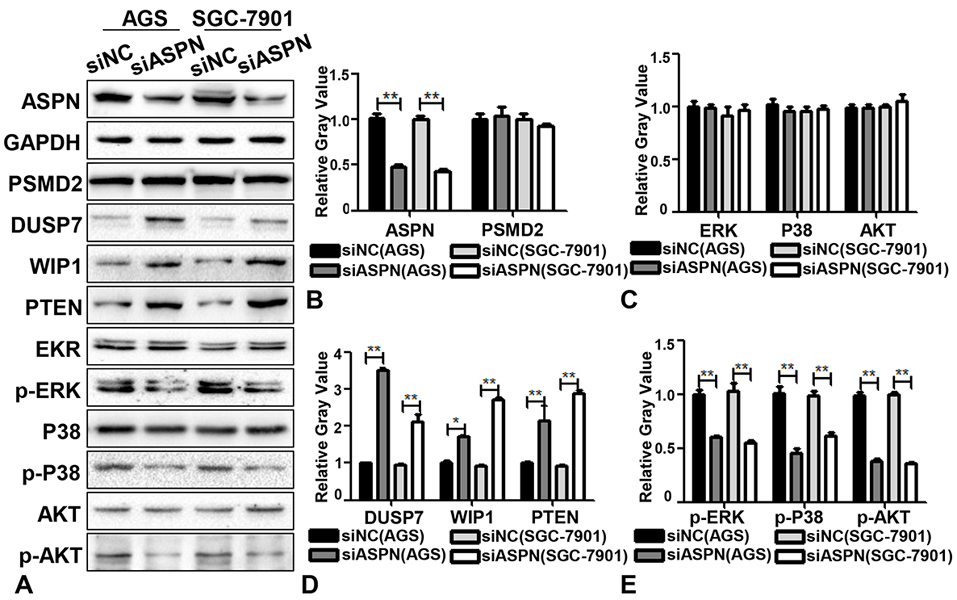 Figure 3.
Figure 3.Knockdown of ASPN promotes the expression of DUSP7, WISP1, PTEN and decreases the phosphorylation of ERK, P38, AKT. (A) At the protein level, the expression of DUSP7, WISP1, PTEN were upregulated and p-ERK, p-P38, p-AKT were decreased when ASPN was silenced in AGS and SGC-7901 cells while the expression of PSMD2, EKR, P38 and AKT has no difference. (B-E) Relative gray value (of control) analysis of ASPN, PSMD2, DUSP7, WISP1, PTEN, ERK, P38, AKT, p-ERK, p-P38 and p-AKT by ASPN knockdown in AGS and SGC-7901. Values are presented as the mean±SD. *P<0.05; **P<0.01.
To further confirm whether PSMD2 is involved in mediating the promotion of proliferation by ASPN in GC cells, several rescue experiments were designed. AGS cells stably expressing ectopic ASPN were transfected with siPSMD2 for 48 h, and WB analysis was subsequently performed. We first verified that two parallel siRNA sequences of PSMD2 were effective enough to knock down the expression of PSMD2 (Figure 4A). According to the knockdown effects of PSMD2, we applied siPSMD2 (2) in the following experiments. The quantitative results of the rescue WB assays demonstrated that ectopic ASPN could reduce the expression of DUSP7, WIP1 and PTEN and induce the phosphorylation of P38, ERK and AKT, while siPSMD2 significantly reversed the expression of the above molecules to baseline levels (Figure 4B-F). In addition to the fact that PSMD2 could interact and colocalize with ASPN, we concluded that PSMD2 mediated the regulatory effects of ASPN on DUSP7, WIP1 and PTEN signaling. Taken together, our study revealed that ASPN could promote cell proliferation by interacting with PSMD2 in GC cells, thereby possibly leading to enhancement of the ubiquitin-proteasome system and facilitating the phosphorylation of ERK, P38 and AKT (Figure 5A).
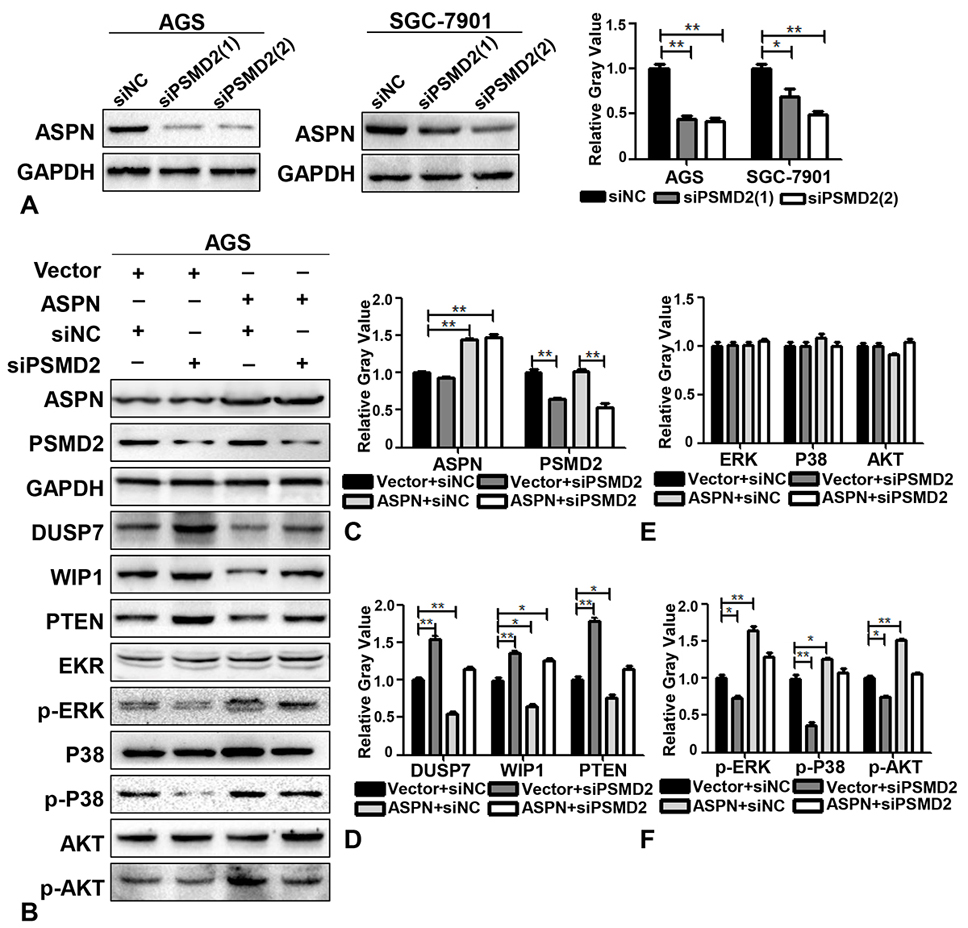 Figure 4.
Figure 4.ASPN promotes the degradation of DUSP7, WISP1, and PTEN and the phosphorylation of ERK, P38, and AKT via PSMD2. (A) WB results and the relative gray value (of control) analysis of the off-target effects of two siPSMD2 sequences in AGS and SGC-7901. (B) The expression levels of GAPDH, ASPN, DUSP7, WISP1, PTEN EKR, p-ERK, P38, p-P38, AKT and p-AKT were detected by WB after cotransfection of AGS cells with empty vector+siNC, empty vector+siPSMD2, ectopic ASPN+siNC and ectopic ASPN+siPSMD2. (C-F) Relative gray value (of control) analysis of ASPN, PSMD2, DUSP7, WISP1, PTEN, ERK, P38, AKT, p-ERK, p-P38 and p-AKT by ectopic ASPN and/or siPSMD2 in AGS cells. Values are presented as the mean±SD. *P<0.05; **P<0.01.
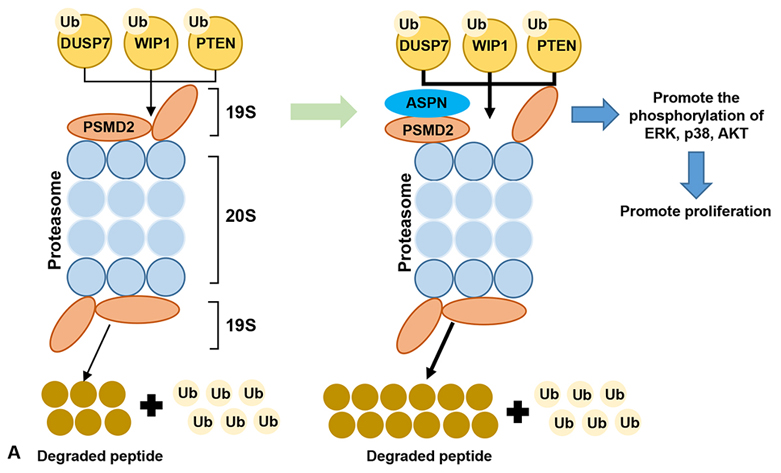 Figure 5.
Figure 5.Schematic shows that ASPN could promote the proliferation of GC by promoting the degradation of the tumor suppressor factors (DUSP7, WIP1 and PTEN), and then facilitate the phosphorylation of ERK, P38 and AKT via interacting with PSMD2 mediated ubiquitin-proteasome system.
ASPN is a secretory protein found in the extracellular matrix (ECM). Previous studies have shown that the polymorphism of the aspartic acid repeat region in ASPN has a notable association with osteoarthritis (10). A few studies have recently revealed that in the tumor microenvironment, CAF-derived ASPN can promote the epithelial to mesenchymal transition (EMT) process of cancer cells (12-17). Although Ding et al. reported the higher expression of ASPN in tumor tissue and the general tumor promoting effect of ASPN inside cancer cells, the exact function (such as the effect on cell proliferation, cell death, cell survival, etc.) of intracellular ASPN and its underlying mechanism in GC have not been completely understood in view of the involvement of the KRAS mutation in EGFR signaling in the AGS cell line. Via a series of EdU and MTS assays for knockdown and overexpression of ASPN, we report for the first time that intracellular ASPN can promote the proliferation and cell viability of GC cells. Mass spectrometry after immunoprecipitation and coimmunoprecipitation assays revealed and verified that endogenous ASPN had strong binding to endogenous PSMD2 in AGS cells. Further immunofluorescence studies showed that PSMD2 and ASPN were colocalized in the cytoplasm and nuclei. Taken together, we demonstrated for the first time that ASPN plays an important role in GC cell proliferation and can interact with PSMD2 in GC cells.
Our WB analysis demonstrated that knockdown of ASPN increased the protein levels of DUSP7, WIP1 and PTEN and decreased the phosphorylation of ERK, P38 and AKT. All three of the above pathways (MAPK/ERK pathway, P38 MAPK pathway and AKT/PKB signaling) have been confirmed to be involved in the regulation of cell proliferation (33, 34). The activation of these pathways (such as the mutation of some upstream molecules) results in the phosphorylation of ERK, P38 and AKT and then enhances their kinase activity, thereby leading to phosphorylation of their downstream targets, which could directly regulate cell proliferation. During the past decades, DUSP7, WIP1 and PTEN have been identified as inhibitors of their own signaling; for example, DUSP7 inhibits RAS-ERK signaling transduction, WIP1 dephosphorylates P38 MAPK, and PTEN represses PI3K-AKT signaling (35-37). Thus, we concluded that ASPN might promote the phosphorylation of ERK, P38 and AKT by enhancing the degradation of DUSP7, WIP1 and PTEN. To further explore whether PSMD2 is involved in the regulation of ASPN on DUSP7, WIP1 and PTEN, we performed a rescue WB analysis via ectopic ASPN overexpression and siPSMD2. The regulatory effects of ectopic ASPN attenuated by siPSMD2 confirmed that PSMD2 mediated the regulating effects of ASPN expression on DUSP7, WIP1, and PTEN and their downstream factors.
Proteasomes are known as the main component of the ubiquitin-proteasome system and the key regulator in cell proliferation via the degradation of DUSP7, WIP1 and PTEN, and they promote the phosphorylation of ERK, P38 and AKT in physiological processes and tumorigenesis (22, 23, 38). As a key subunit of the 19S regulator lid, PSMD2 was reported to promote the binding ability and the degradation function of proteasomes (21). In addition, recent studies identified p-ERK, p-P38 and p-AKT as potential downstream effectors of PSMD2, which could be deregulated by knocking down the expression of PSMD2 in lung cancer and breast cancer (28, 29). Taken together, we hypothesized that the interaction between ASPN and PSMD2 might enhance the degradation of these tumor inhibitor factors via the ubiquitin-proteasome system and activate the corresponding signaling pathway to induce tumorigenesis. Although we confirmed that PSMD2 mediated the regulatory effect between ASPN and downstream factors, the specific molecular mechanism of the protein structural interaction between ASPN and PSMD2 and how the interaction enhances degradation of the ubiquitin-proteasome system need to be further explored.
Since the late 1990s, inhibition of proteasome function has emerged as a powerful strategy for anticancer therapy. The first two generations of proteasome inhibitors have dramatically improved outcomes in patients with myeloma and other cancers; however, frequent relapses and resistance to treatment have restricted the further utility of these drugs (31). Although novel proteasome inhibitors have been recently identified, such as FGD5 (which sustains VEGFA signaling by inhibiting VEGFR2 degradation) and MG132 (which systemically perturbs the ERK signaling pathway), the clinical effects of these inhibitors remain unclear, and the frequency of relapses and resistance need to be further studied (39, 40). Our study sheds new light on the combined application of ASPN targeting therapy and proteasome inhibitors in antitumor treatment; moreover, this combined therapy may reduce drug resistance.
In summary, we revealed for the first time that ASPN is located inside GC cells and promotes the proliferation of GC cells by interacting with PSMD2, thereby promoting the degradation of DUSP7, WIP1 and PTEN. P-ERK, p-P38 and p-AKT signaling are potential downstream targets of the ASPN-PSMD2 axis. Our findings also suggest that ASPN and its downstream molecules could serve as potential therapeutic targets for GC. The combined application of ASPN targeted therapy may enhance the antitumor effect of proteasome inhibitors and reduce drug resistance.
Zheng Zhang, Hengcun Li, equally contributed to this paper. This study was supported by National Natural Science Foundation of China (81702314, 81570507) and National Key Research and Development Program of China (2017YFC0113600).
Abbreviations: ATCC: American Type Culture Collection; ASPN: asporin; GC: gastric cancer; CAFs: cancer-associated fibroblasts; ECM: extracellular matrix; EMT: mesenchymal transition; FBS: fetal bovine serum; PSMD2: 26S proteasome non-ATPase regulatory subunit 2; SLRP: small leucine-rich proteoglycan; PBS: phosphate-buffered saline