
Experimental evidence has shown that chimeric switch receptor T (CSR-T) cells, activated by binding programmed death-ligand 1 on the tumor cell surface, lead to tumor regression in experimental animals. In this phase I clinical study, we evaluated the safety and bioactivity of CSR-T cell therapy in 14 patients with recurrent glioblastoma who were unresponsive to surgical resection and standard radiotherapy. Patients who received 108 CSR-T cells either intravenously or intracranially showed an increase in the levels of IFN-gamma and IL-6, respectively, in peripheral blood or cerbrospinal fluid (CSF). Moreover, the number of T cells present in CSF significantly increased after the treatment. Patients did not show grade 3 or 4 adverse effects. The evidence of in vivo biological activity and lack of adverse effects of treatment with CSR-T cells suggest that such treatment can be subjected to further analysis to show the efficacy of this new treatment strategy in the treatment of cancers that are not responsive to traditional therapeutic regimens.
Glioblastoma (GBM) is the most common and deadliest malignant primary brain tumor. Despite the multimodal therapeutic approaches, including surgical resection, radiotherapy, and chemotherapy, GBM demonstrates a high recurrence rate with poor patient prognosis. Thus, the median survival time of patients with GBM is as low as 12–15 months, with a 5-year survival rate of <10% (1).
Conventional therapies for GBM cannot be directed against tumor cells alone, and their non-specific toxicity limits their usage. Therefore, immune targeting of tumor-specific gene mutations may better eradicate tumor cells. Anti-CD19 chimeric antigen receptor T (CAR-T) cell therapy has achieved remarkable results in the treatment of relapsed and/or drug-resistant hematological cancer, such as refractory B-cell leukemia and lymphoma (2, 3). However, extensive clinical treatment of solid tumors, including brain tumors, is complex and currently under in-depth investigation. Recently, Brown et al performed multiple intraventricular infusions of CAR-T cells targeting IL13Ra2 in a 50-year-old patient with recurrent GBM. The procedure resulted in the regression of all intracranial and spinal tumors with an increase in the cytokine levels and immune cell number in cerebrospinal fluid (CSF) for over 7.5 months (4). This suggests that the local delivery of CAR-T cells enhances the immune response to tumor cells. Moreover, different administration methods may have distinct therapeutic effects (5).
PD1 is a key immune molecule that prevents T cell immune hyperactivity and autoimmune diseases, and its ligand, programmed death-ligand 1 (PD-L1), is widely expressed in GBM (88% of cases) and other tumors. Therefore, anti-PD-L1 or PD1 antibody immunotherapy is likely to achieve significant anti-tumor effects in GBM. Both PD1 and CD28 are members of the CD28 family of proteins (6). T cell activation requires not only binding of antigens to T cell receptor (TCR) to provide activation signals but also co-stimulatory molecules to provide immune signals. CD28 is the major co-stimulatory receptor for CD4+ helper T cells and CD8+ cytotoxic T lymphocyte cells.
We previously constructed an anti-human PD-L1 chimeric receptor that not only recognizes tumor cells but also converts signals that inhibit T cell activity into those that induce T cell activation (7). Interestingly, the chimeric protein, which encompasses the extracellular domain of CD28 and the intracellular region of PD1, exhibits an activity similar to that of the native PD1 molecule in inhibiting the T cell immune response (7). The chimeric protein is capable of binding to PD-Ll and promoting T cell activation, with increased cytokine release, cell proliferation, and cell cytotoxicity (8, 9). Injection of T cells expressing PD1-CD28 chimeric receptors into tumor-bearing mice completely inhibited tumor growth (Figure 1) (9). These findings indicated that the different functional regions of these two proteins are compatible and can be combined to form a new artificial receptor capable of modulating the T cell immune response. Interestingly, the PD1-CD28 chimeric receptor induces PD-L1/PD1 signaling, which usually inhibits T cell activity and converts it into a signal inducing T cell activation. Thus, similar to CAR-T cell therapy, gene-modified chimeric switch receptor T (CSR-T) cell therapy targeting PD-L1 may achieve anti-tumor effects. CSR-T cells simultaneously express an emergency brake system (EBS). EBS, when activated, can immediately clear CSR-T cells, preventing excessive immune reactions.
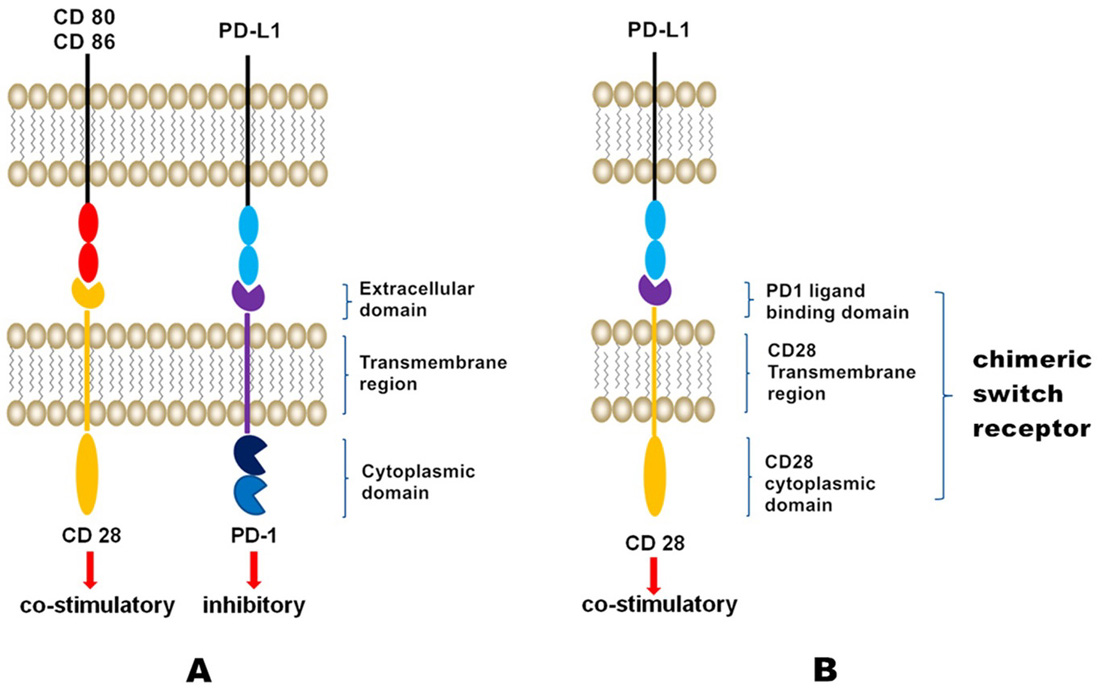 Figure 1.
Figure 1.
Schematic of CSR-T cell technology. The extracellular region of PD1 and the transmembrane and membrane regions of CD28 were fused to form a chimeric protein capable of binding to PD-Ll and promoting T cell activation, with increased cytokine release, cell proliferation, and cell cytotoxicity.
In the present study, CSR-T cells were constructed from autologous peripheral blood T lymphocytes using the lentiviral vector and were employed to treat PD-L1-positive GBM patients. The safety and bioactivity of CSR-T cell therapy targeting the PD-L1 were evaluated.
This was an open-label, uncontrolled, single-center phase I clinical trial performed to evaluate the feasibility, safety, and bioactivity of CSR-T cell therapy in 14 patients with recurrent GBM. The study was performed in a hospital setting, and written informed consent was obtained from all included patients. The clinical protocol was approved by the Institutional Review Board of SanBo Brain Hospital, Capital Medical University (SBNK-2016-016-01; Beijing, China). The study has been registered at ClinicalTrials.gov (NCT02937844). Beijing Marino Biotech Co., Ltd. provided technical support for the preparation of CSR-T cells as well as flow cytometry and PCR.
Eligible patients were adults (aged 18–70 years) with recurrent or refractory GBM, who had undergone surgical resection as well as standard radiotherapy and routine chemotherapy. The tumor samples were determined to be PD-L1-positive by immunohistochemistry (Figure 2). Prior to enrollment, patients were required to be steroid independent, to have completed primary therapy (≥2 weeks), and to be recovering from all acute side effects. Other inclusion criteria included: normal liver and kidney function; normal bone marrow function; the absolute number of peripheral blood lymphocytes ≥ 0.8×109/L; absolute neutrophil count ≥ 1.5×109/L; platelet count > 100×109/L; hemoglobin > 100 g/L; negative pregnancy test for females; and absolute count of peripheral blood lymphocytes ≥ 0.8×106. Patients with active infection, unexplained febrile illness, unstable or severe medical conditions (e.g., severe heart or lung disease or active hepatitis), inflammatory conditions such as rheumatoid arthritis, known immunosuppressive disease or human immunodeficiency virus infection, or treatment with corticosteroids above physiologic doses (>2 mg/d dexamethasone) during CSR-T cell immunotherapy were excluded.
 Figure 2.
Figure 2.
PD-L1 staining in tumor cells. PD-L1 is localized in the cell membrane and cytoplasm of GBM tumor cells. IHC was used for analysis. Negative expression of PD-L1 in GBM tumor cells (A); positive expression of PD-L1 staining in GBM tumor cells (B,C) (original magnification, 200×)
The sections of deparaffinized and rehydrated tumor tissue samples were subjected to antigen retrieval via microwave heating method using EDTA (at a pH of 8.0 and concentration of 1mmol/L), followed by blocking of endogenous peroxidase activity. Samples were then treated with 100–200 µl of PD-L1 primary antibody (CST 13684#, 1:200, blocking solution) and incubated overnight at 4°C. On the following day, samples were equilibrated at room temperature (RT) for 10–15 min, washed 3 times (5 min each) in 1× PBS, and incubated with secondary antibody (Life A16110) for 1 h at RT. Following a wash, the samples were stained with DAB for 10 min, washed again, and stained for nucleus with hematoxylin; a coverslip was then placed. Under the light microscope, ten random fields were examined, in which the staining (cytoplasm or membranes) in over 5% of tumor cells was considered PD-L1 positive (10).
At enrollment, each patient underwent leukapheresis for collection of peripheral blood mononuclear cells, which were used to engineer CSR-T cells. Subsequently, confirmed therapeutic CSR-T cells were cryopreserved and stored for later use. Before use, CSR-T cells were thawed and re-expanded in vitro using a rapid expansion method.
Prior to infusion, the CSR-T cells were preconditioned with 250 mg/m2 d1-3 of cyclophosphamide. Then, 14 GBM patients, who met all the study criteria, received up to three consecutive intravenous infusions of CSR-T cells (maximum dose of 109 cells) at intervals of 4 days. The planned transfusion of CSR-T cells was divided into three phases. For safety, the CSR-T cell dose was gradually increased at each consecutive infusion. Intravenous infusion of CSR-T cells was followed by infusion of saline, both of which were conducted slowly (about half an hour per treatment). Each patient’s response, heart rate, and blood pressure were monitored.
One of the patients received up to three consecutive intravenous infusions at a maximum dose of 108 CSR-T cells and four local intracranial infusions at a maximum dose of 106 CSR-T cells via the catheter/reservoir system. Patients did not undergo any routine pretreatment, such as chemotherapy, prior to treatment with CSR-T cells. This patient was selected because he met the administration criteria; the patient had recurrent glioblastoma, even after multiple operations, radiotherapy, and chemotherapy, with occurrence of multiple intracranial dissemination. The three doses of CSR-T cells were delivered weekly for 3 weeks, and the fourth dose was given after a 2-week interval (in the fifth week), both through the catheter/reservoir system. The catheter/reservoir system was installed in advance, wherein one end of the catheter was placed into the tumor cavity, whereas the reservoir end was installed on the scalp, facilitating an intracranial passage into the tumor cavity; 30 minutes before the transfusion, the patient was treated with 15 mg/kg of acetaminophen per os (max dose 650 mg) and 25–50 mg diphenhydramine IV or per os (max dose 50 mg). Using a 21-gauge butterfly needle, 0.5 ml of cell suspension was slowly placed into the chemotherapy sac (5–10 minutes) pushed into the tumor cavity. This was followed by infusion of 2 ml saline with an infusion pump, at a rate of 1 ml/hour, to flush the remaining cells in the drainage tube. Cells were returned via return injection each time through an intracranial CSR-T cell transfusion capsule, that is the catheter/reservoir system. The dosages of CSR-T cells in the return injection were 5.0×105, 1.5×106, and 3.0×106 CSR-T cells in the first, second, and third weeks, respectively. After transfusion, the patient was observed for at least 3 hours, and vital signs were measured, including the heart rate, blood pressure, respiration, body temperature, KPS score, and other clinical parameters. At week 5, 4.0×106 CSR-T cells were injected into the tumor cavity via the catheter/reservoir system.
Patients with intracranial drug administration were considered eligible for intravenous administration. The patient who received intravenous infusion also received intracranial administration after a 2-week interval. CSR-T cells were prepared with a protective agent and frozen in liquid nitrogen. The frozen cells were thawed, washed with PBS, and stained with trypan blue. More than 80% of living cells were retrieved. For each intracranial administration, we first extracted 5 ml of CSF and then gradually injected the prepared cells into the chemotherapy capsule. Next, we used a pump to slowly inject 2 ml PBS into the chemotherapy sac within 2 hours. CSF samples were taken 2 days after each intracranial injection of CSR-T cells for analysis; Cycle1: intracranial dose 5.0×105, Cycle2: intracranial dose 1.5×106, Cycle3: intracranial dose 3.0×106, and Cycle4: intracranial dose 4.0×106. Cells were isolated from the collected CSF sample and frozen at −80°C. After DNA extraction, the CSR gene copy number was quantified with the Lenti-X qRT-PCR Titration Kit (Clontech) and normalized to that of LX23.
All adverse events (AEs) were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events Version 4.0.3. Level 1 AEs are mild and not life-threatening, and the symptoms include fever, nausea, fatigue, headache, and muscle pain. For these reactions, symptomatic treatment is provided, for example, physical cooling. Level 2 AEs are moderate, with mild respiratory symptoms, grade 2 organ toxicity, or less severe hypotension. These symptoms can be improved by rehydration and a low-dose booster of oxygen (<40%). In the absence of multiple complications in non-elderly subjects showing ≥2 level 2 AEs, therapies can be used to support preventive treatment, with close monitoring of heart and other organ functions. However, in case of multiple level 2 AEs in elderly subjects, treatment should be performed depending on the severity of the symptoms, and whether the IL-6 receptor antagonist (tocilizumab; 8 mg/kg) is required to prevent symptoms from worsening should be assessed. Level 3 AEs are severe, whereas level 4 AEs are life-threatening and include grade 4 organ poisoning, which requires mechanical ventilation.
Based on the working principle of CSR-T cells, we hypothesized that AEs occurring after infusion include cytokine release syndrome (CRS), similar to immune-related AEs in CAR-T cell and anti-PD-L1/PD1 antibody therapies (11-14). CRS occurs owing to the release of cytokines by the transfused T cells, in vivo, in response to tumor antigens. CRS may occur from days 2 to 15 after infusion and was graded according to the CRS grading system criteria (15). For grade 1 and 2 events, including a headache, fatigue, myalgia, olfactory auras, and neurological symptoms, treatment measures included dexamethasone administration, activation of EBS, and use of Erbitux® (cetuximab; 400 mg/m2 IV within 2 hours). In subjects with grade 3 and 4 AEs, 8 mg/kg IL-6 receptor antagonist (Yamero ®/tocilizumab; Harother & Co., Ltd.) was administered to relieve symptoms immediately. If no improvement was seen within 24 hours of tocilizumab administration, hormone drugs, such as methylprednisolone (2 mg/kg/day) or dexamethasone (0.5 mg/kg; maximum dose of 10 mg/dose), were considered.
Short-term efficacy was evaluated according to the MacDonald standard and judged based on the iRANO standard in case of false progression. Complete remission (CR) was defined by the following criteria: the disappearance of enhanced lesions for at least 4 weeks without new lesions; stabilization or improvement of T2/fluid-attenuated inversion recovery (FLAIR) non-enhanced lesions; and clinical signs remaining stable or improving without the use of corticosteroids. Partial remission (PR) was defined by the following norms: lesion size reduced by 50% for at least 4 weeks and no recurrence; stabilization or improvement of T2/FLAIR non-enhanced lesions; stable or reduced corticosteroid dose; and stable or improved clinical signs. Progressive disease (PD) or disease recurrence was defined by: increase in lesion size by 25%; formation of a new lesion; significantly worsened T2/FLAIR non-enhanced lesions; overt appearance of decreased clinical signs; and stable or reduced corticosteroid dose. Stable disease (SD) was defined by: not meeting the CR, PR, or PD criteria, for example, the total lesion size decreased or increased but not up to that in the PR definition or no new lesions. Time to survival was assessed from CSR-T cell infusion and initial histologic diagnosis to death or the last follow-up.
Plasma IFN-γ and IL-6 levels were measured by ELISA for evaluating the bioactivity of CSR-T cells. PCR was used to detect DNA copy numbers for accessing the amplification of CSR-T cells.
All data were analyzed using SPSS 21.0 statistical software (SPSS Inc., Chicago, IL, USA). To detect changes in IFN-γ and IL-6 levels before and after infusion of cells, t-tests were used. Descriptive statistics were used to assess quantitative variables, for example, mean and standard deviation or median and interquartile range. The median survival time and related 95% CIs were calculated by the Kaplan–Meier method.
In this phase I trial, 14 patients with supratentorial GBM received up to three consecutive intravenous infusions of CSR-T cells, whereas one of them further received four consecutive intracranial infusions of the same. A detailed summary of the patient population is provided in Table 1. All but one of the enrolled study subjects were males, with their age ranging from 28 to 62 years (median, 40 years). The median survival after histologic diagnosis was 17.00 months (95% CI: 9.67–24.33) (Figure 3). The proportions of patients alive at 1, 3, and 6 months after histologic diagnosis were 1, 1, and 0.93 (95% CI: 0.59–0.99), respectively. The median survival time after intravenous infusion of CSR-T cells was 4.40 months (95% CI: 1.83–6.97).
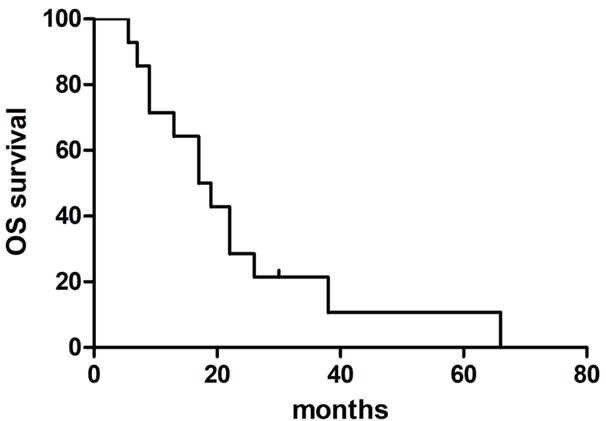 Figure 3.
Figure 3.
Survival analysis. The median overall survival was 17 months (95% CI: 9.67–24.33) in patients treated with CSR-T cells.
Toxicity was minimal, and no AE exceeded grade 2 toxicity at any of the tested intravenous infusion doses of CSR-T cells. Moreover, no CRS or neurotoxic symptom was observed, and all AEs were transient and manageable. Therefore, a maximum tolerated dose was not established, but a practical upper limit for repeated CSR-T cell infusion was set at ≤1.4×109, which was considered safe in all patients. The mean feasible dose was 3.0×108 (SD: 3.67×108) CSR-T cells. One patient received three consecutive intravenous infusions at a maximum dose of 108 CSR-T cells and four local intracranial infusions at a maximum dose of 106 CSR-T cells via the catheter/reservoir system. During the first 3 weeks of post intracranial infusions of CSR-T cells, the patient developed a brief low fever, fatigue, and olfactory auras after each cell administration, which self-resolved within 24 hours. In the fifth week, 32 hours after receiving the maximum dose (4.0×106) of CSR-T cells, the patient showed frequent epileptic events, which resolved after treatment with dexamethasone and sodium valproate.
The plasma IFN-γ and IL-6 levels were assessed using ELISA to evaluate the bioactivity of intravenously administered CSR-T cells in 14 patients who received intravenous infusion at doses of 106, 107, and 108 cells before and after infusion. IFN-γ and IL-6 levels after intravenous infusion of 109 were not assessed. Plasma IFN-γ and IL-6 levels increased with increasing doses of CSR-T cells. The mean plasma concentration of IFN-γ before treatment was 9.13 ± 0.35 pg/ml, which increased to 10.02 ± 0.58 pg/ml (P < .001) after the third intravenous infusion of CSR-T cells (Figure 4). Similarly, the mean plasma concentration of IL-6 increased from 15.09 ± 1.54 pg/ml before treatment to 15.92 ± 1.61 pg/ml (P < .05) after the third intravenous infusion of CSR-T cells (Figure 5).
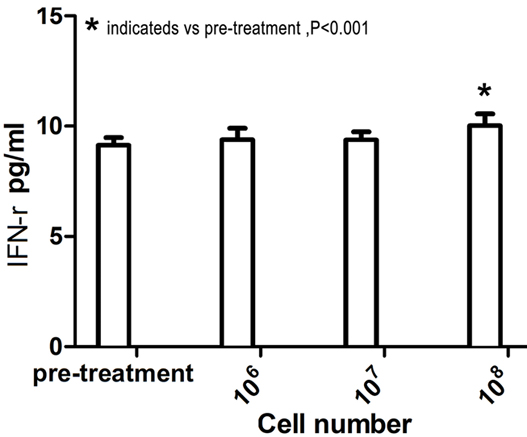 Figure 4.
Figure 4.
Plasma IFN-γ levels increased with increasing doses of CSR-T cells. The abscissa represents the number of CSR-T cells infused intravenously, and the ordinate represents the concentration of IFN-γ in the blood. There was a clear increase in the concentration of IFN-γ in the blood when comparing levels before treatment and after the third intravenous infusion of CSR-T cells (9.13 ± 0.35 pg/ml vs. 10.02 ± 0.58 pg/ml, P < .001).
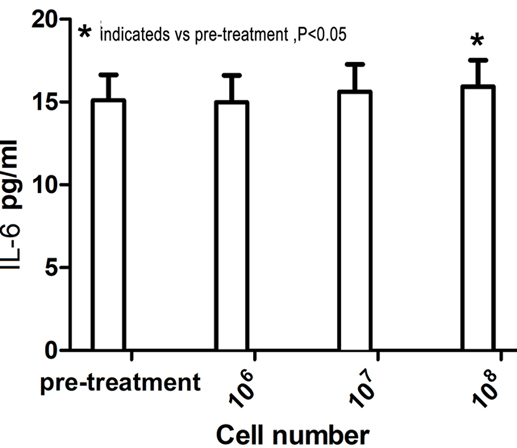 Figure 5.
Figure 5.
Plasma IL-6 levels increased with increasing doses of CSR-T cells. There was a clear increase in the concentration of IL-6 in the blood when comparing levels before treatment and after the third intravenous infusion of CSR-T cells (15.09 ± 1.54 pg/ml vs. 15.92 ± 1.61 pg/ml, P < .05).
Only one patient received a maximum dose of 109 cells via intravenous infusion, whereas all 14 received a maximum dose of 108 cells via intravenous infusion. Owing to the limited number of patients in the trial treated with 109 CSR-T cells, the corresponding data could not be analyzed.
The cells obtained from the collected CSF samples were quantified for CSR gene copy numbers. The results showed that CSR-T cell numbers peaked on the third day after each transfusion. Moreover, a significant increase in CSR gene copy numbers was detected after the third and fourth cell transfusions, indicating that CSR-T cells were activated and amplified in vivo (Figure 6). These results, with the lower dose (106) of CSR-T cells used for intracranial injection, suggest that CSR-T cells exhibit a higher bioavailability following intracranial injection than that following intravenous administration.
 Figure 6.
Figure 6.
Number of CSR-T cells in patient injected intracranially via the catheter/reservoir system. Intracranial administration was performed at doses of 5.0×105 (1st), 1.5×106 (2nd), 3.0 ×106 (3rd), and 4.0×106 (4th). There were no increases in the DNA copy numbers after the first or second administration; however, an obvious DNA copy number increase was observed after the third and fourth administrations.
This study demonstrated the feasibility and safety of consecutive intravenous or intracranial infusions of CSR-T cells targeting PD-L1 in patients with GBM. With no patient experiencing grade 3 or 4 AEs, the procedure was considered safe. Further, a temporary biological activity of CSR-T cells was also observed.
CAR-T cell therapy has demonstrated remarkable clinical success in the treatment of CD19+ hematological malignancies; infact, most patients with acute lymphoblastic leukemia achieve complete remission (2, 16). A major goal in this field is the successful application of CAR-T cell therapy to a wide range of solid tumors. However, achieving this will require overcoming critical barriers to therapeutic success, including tumor antigen selection, tumor heterogeneity, and the immunosuppressive tumor microenvironment. The current study focused on the development of CSR-T cell therapy, which is similar to CAR-T cell therapy, and investigated its utility in GBM treatment. Resolving this type of tumor by existing standard treatment options is hard. Preclinical studies demonstrated that CAR-T cells display potent anti-glioma responses in orthotopic mouse models (17, 18); thus, CAR-T cell immunotherapy may improve the treatment of GBM. A major design challenge for glioma immunotherapy is the identification of tumor-specific target antigens. Promising cell surface markers that are being pursued for the development of CAR-T cell therapy for high-grade gliomas include IL13Rα2, HER2, EGFRvIII, and EphA2 (18-21). However, the number of patients with GBM expressing these markers is low; for example, EGFRvIII is found in approximately 30% of the total glioblastoma cases (22). In contrast, PD-L1 is overexpressed in >88% GBM cases, whereas it is not expressed in healthy brain tissue (23).
Activated, but uncontrolled, T cells are highly destructive and can severely damage normal tissues. The PD1 receptor, by inhibition of TCR-transduced T cell activation signaling, plays a key role in preventing self-reaction of T cells and autoimmunity. Activation of PD1 signaling can also disable anti-tumor immune reactions, leading to tumor immune escape. Overexpression of PD-L1 is observed in several tumor types. PD-L1/PD1 signaling is one of the many strategies employed by tumors to escape immune surveillance. Antibodies, either by binding to PD1 or PD-L1, block the binding of PD-L1 to PD1, thus inhibiting PD1 signaling and reactivating the anti-tumor response. Multiple trials have demonstrated that immune checkpoint blockers, such as nivolumab, pembrolizumab, atezolizumab, and durvalumab, have significant survival benefits over conventional therapies in several types of recurrent or refractory malignancies. Recently, trials have been conducted to evaluate the safety and efficacy of immune checkpoint blockers in GBM, with a variety of approaches, including immune checkpoint blockade therapy alone or in combination with another therapeutic option (e.g., bevacizumab chemotherapy and radiotherapy).
The anti-human PD-L1 chimeric receptor can identify and eliminate PD-L1-positive tumor cells. The PD1-CD28 chimeric receptor induces PD-L1/PD1 signaling, which usually inhibits T cell activity, and converts it into a signal that stimulates T cell activation. It simultaneously expresses an EBS to immediately clear CSR-T cells in case of emergency. EBS involves a truncated, nonfunctional EGFR (tEGFR) (24), which is devoid of the original EGFR ligand binding domain as well as the intracellular receptor activation region, but retains the transmembrane and recognition regions of the anti-EGFR antibody (24). In case of excessive life-threatening toxicity during the course of treatment, FDA-approved anti-EGFR monoclonal antibodies, such as Erbitux®, can be used to cleave T cells expressing chimeric receptors (24). In the vector, tEGFR, containing an IRES for translation, was inserted upstream of the CSR gene, and the entire sequence was cloned into the lentiviral vector. CSR and tEGFR were simultaneously transfected into T cells using a lentiviral vector. The expression of tEGFR did not affect CSR cytotoxicity, and anti-EGFR antibodies were effective in cleaving T cells expressing tEGFR. This ensures the safety of CSR-T cells in vivo.
This clinical study involving the treatment of 14 patients revealed that PD-L1 is a suitable target molecule for CSR-T cell immunotherapy. It established patient tolerance and acceptable safety profiles for gradual increases in intravenous infusions of CSR-T cells with transient and manageable AEs. Preliminary results demonstrated that anti-CSR-T cell therapy was safe and feasible in GBM patients without CRS or neurotoxicity. In addition, after IV infusion of CSR-T cells, the levels of IFN-γ and IL-6 in the peripheral blood increased with gradual increases in the number of reinfused cells (up to 109), indicating a biological activity of CSR-T cells. After intracranial injection of CSR-T cells, higher levels of CSR-T cell proliferation were detected in CSF at a lower injection dose (106 cells). Thus, immunological evidence indicates that CSR-T cells were activated and amplified in the patient. Together, these findings indicate that the local intracranial injection of CSR-T cells tends to be more effective than intravenous reinfusion, and this is consistent with results reported by Brown et al (25).
It should be pointed out that the sample size of this study was limited, as was the significance of the results. However, because this was a phase I clinical trial, it was necessary to explore the safety of CSR-T cells and the optimal dose of injected cells before conducting a study with a large sample size. We plan to increase the sample size and dose of cells for peripheral intravenous infusion. More importantly, we hope to further investigate the utility of intracranial injections via the recruitment of additional patients, so that we can further explore the safety and biological activity of CSR-T cells.
In conclusion, our results demonstrated that continuous intravenous infusion of CSR-T cells is safe in patients with GBM and leads to a temporary biological activity of CSR-T cells. Moreover, the intracranial local administration may result in improved biological activity. These findings provide unprecedented clinical results for GBM treatment by CSR-T cell therapy and lay the foundation for the improvement of CSR-T cell therapy in the future.
The authors would like to thank the investigators, patients, and their families. We are also grateful to Beijing Malary Biotech Co., Ltd. for its support.
Abbreviations: AEs: adverse events; CAR-T: chimeric antigen receptor T; CSR-T: Chimeric switch receptor T; FLAIR: fluid-attenuated inversion recovery; GBM: Glioblastoma; PD-L1: Programmed death-ligand 1; RT: Room temperature; TCR: T cell receptor; IFN-γ: Interferon-gamma; IL-6: Interleukin- 6; EGFR: Epidermal growth factor receptor; tEGFR: Truncated EGFR; EBS: Truncated, nonfunctional EGFR