
Vascular remodeling is a critical event following a stroke. It is a well known fact that C1q is the first molecule in the complement classical pathway. However, its role in the neovascularization that ocurs after a stroke, remains unclear. In this study, we investigated the effects of C1q on post-stroke angiogeneis in pMCAO rats. We found that increased C1q levels in IBZ enhanced angiogenesis in rats with pMCAO. C1q promoted viability and angiogenic function of RBMECs and HBMECs. Upregualtion of VEGF expression and secretion by C1q was also observed in RBMECs, HBMECs and in IBZ in pMCAO rats. Furthermore, we demonstrated that C1q enhanced angiogenic function of RBMECs through its receptor, LAIR1. The results show that administration of C1q enhanced neovascularization and reduced brain edema after pMCAO in rats. On the basis of these findings, we suggest that C1q plays an important role in post-stroke angiogenesis at least through LAIR- HIF1α-VEGF axis. C1q shows promise as a potential therapeutic candidate for stroke treatment.
C1q is part of the C1 complex, the first component of complement in classical complement pathway. C1q serves to recognize signals that lead to sequential proteolytic activation of complement classical pathway in innate immune system (1). C1q is formed from 18 peptide chains in 3 subunits of 6 chains. A collagen-like tail is located near the N terminal and a globular region resides at the C terminal (2). Not only antibody-dependent but also independent immune functions have been assigned to C1q. Further, C1q functions are not just restricted to the immune system. Recent studies demonstrate that C1q plays an important roles in activation of tumor suppression in prostate cancer (3), placenta development (4), and wound healing in chronic skin ulcers in diabetic patients (5). Functions of C1q are considered to be mediated by C1q receptors (2). LAIR1, one of C1q receptors, is a collagen receptor that is involved in regulation of dendritic cell differentiation and NK cell activation by C1q (6).
LAIR-1 is a transmembrane protein in Ig superfamily (6). LAIR1 extracellular domain can bind to collagen tail of C1q in a hydroxyproline-dependent fashion (6). LAIR1 intracellular domain is composed of two immunoreceptor tyrosine-based motifs that can be phosphorylated to negatively regulate signaling associated with immune cell functions (7-10). So far, the role of C1q/LAIR1 signaling in recovery of stroke has being poorly defined.
Vascular remodeling is a critical event after a stroke, which is found to occur in the areas of new neuroblasts (11). Accumulated evidence has successfully proved that angiogenesis occurs in patients and animals after brain ischemia, and that angiogenesis is critical for recovery after the stroke (12, 13). Newly formed blood vessels, chemokines and growth factors secreted from microvascular endothelial cells play a pivotal role in supporting the survival of new neurons (14).
Recent studies demonstrated that C1q is associated with angiogenesis in wound healing during the treatment of chronic skin ulcers in diabetic patients (5), and also under hypoxia conditions after brain injury (15). However, role of C1q in angiogenesis after stroke still remains uncertain. In this study, we investigated the effect of C1q on angiogenesis in pMCAO rats and explored the involvement of LAIR1 in the molecular mechanisms underlying the regulation of angiogenesis after the stroke.
All animal experiments were carried out in compliance with regulations of the institutional committee of animal care and use. The animal protocol was approved by the medical ethics committee of Nanjing University School of Medicine, Nanjing, P.R.China. Adult male Sprague Dawley rats were maintained in an animal facility at Nanjing Medical University, China. Rats were anesthetized using isoflurane. PMCAO was performed as previously described (16). To assess the effect of C1q, lateral ventricular injection of 50 μg C1q α, β and γ siRNA mixture (Santa Cruz Biotech, USA) or 5 mg/kg full length native C1q (Abcam, USA) was given 1 h after pMCAO.
Whole cell extracts were separated by 4-12% SDS-PAGE (Bio-Rad, USA) and transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore, USA). The membrane was then blocked with 5% nonfat milk in TBST (10 mM Tris, pH 8.0, 150 mM NaCl, 0.5% Tween 20) for 1 h. After washing with TBST, the membrane was incubated with antibodies against C1q (1:1000) (Abcam, USA), HIF-1α (1μg/ml) (R&D Systems, USA), VEGF (1:1000) (Novus Biologicals, USA), LAIR1 (1:1000) (Abcam, USA) or β-actin (2 μg/ml) (Boster Biological Technology, USA) at room temperature for 1 h. The membranes was washed three times for 5 min each and incubated with horseradish peroxidase (HRP)-conjugated anti-rabbit or anti-mouse antibodies (1:10000) (Boster Biological Technology, USA) for 1 h. The membrane was then washed with TBST three times for 10 min each and the blots were developed with the ECL western blot substrate (Promega, USA) according to the manufacturer’s protocols.
Blood vessels were stained with FITC-conjugated anti-CD31 antibody (Bio-Rad, USA) in cryosections as previously described with modifications (17, 18). Nuclei were counterstained with DAPI (Thermo Fisher Scientifc, USA). Staining of capillaries was analyzed under ZEISS Axio Imager M1 fluorescence microscope. Six fields from each section were collected from the IBZ. Quantification of the number of vessels stained with FITC anti-CD31 antibody per 6 high-power fields were proceeded for statistical analysis.
RBMECs and HBMECs (ScienCell, USA) were grown in Endothelial Growth Medium (EGM) from LONZA supplemented with 2% FBS and bovine brain extracts. The cells were all incubated at 37°C in 5% CO2.
Hypoxic culture condition was created in an airtight plexiglas chamber (Bellco Glass, Vineland, NJ). The chamber was deoxygenated by infusion of a 2% O2 and 98% CO2-N2 gas mixture. The chamber containing cultures was then placed in a standard humidified tissue incubator. Control cultures were placed in normoxia. Cells were harvested at 48 h after hypoxia treatmen for western blot analysis.
RBMECs and HBMECs were seeded into a 96-well culture plate (Corning, USA) at a density of 5 × 104 cells/well. 24 h after seeding, the cells were grown in serum free condition in the presence of C1q. 48 h later, the cell viability was examined by using a MTT assay kit (Sigma, USA) according to the manufacturer’s instructions. Optical density (OD) values were read by using a microplate reader (SpectraMAX, Molecular Devices, USA) at an absorption wavelength of 570 nm.
RBMECs and HBMECs were grown to confluence in a 24- well tissue culture plate (Nunc, USA), and the cells were then incubated with increasing concentrations of C1q (1, 10, 50, and 100 μg/ml) for 12 h at room temperature in a serum free condition. Levels of VEGF in culture supernatant were analyzed with a rat VEGF ELISA kit (R&D systems, USA) and a human VEGF ELISA kit (Takara Bio, Japan) respectively.
5 × 105 RBMECs or HBMECs were seeded onto 8-µm Transwell inserts (Corning) in a 24 well culture plate. The lower chamber was filled with 500 μl 2% serum cell culture media. The cells were incubated for 12 h in the presence of 10 μg/ml C1q at 37ºC in incubator. Migrated cells were fixed with 4% formaldehyde, stained with 0.5% crystal violet and examined under Nikon microscope 3. For VEGF blocking experiment, the cells were pretreated with 100 ng/ml VEGF neutralizing antibody (R&D Systems, USA) for 1h.
200 ul Matrigel from BD Biosciences was added to each well of a 24-well plate and incubated at 37ºC for 1 h. 1 × 104 RBMECs or HBMECs were seeded in each well and incubated for 24 h at 37ºC in the presence of 10 μg/ml C1q. Tube formation was assessed and quantified. For VEGF blocking experiment, the cells were pretreated with 100 ng/ml VEGF neutralizing antibody for 1 h followed by C1q treatment.
RBMECs at 80–90% confluence were transfected with 100 nM LAIR1 siRNA or scrambled control siRNA (Thermo Fisher Scientific, USA) by using RNAiMAX transfection reagent (Thermo Fisher Scientific, USA) according to the manufacturer’s instruction. 48 hours after transfection, the cells were treated with C1q for 12 h followed by western blot analysis.
5 days after pMCAO, brain tissues were immediately cut into 4 mm blocks, weighed and heated at 100ºC overnight. The following formula was used to calculate the brain water content: (wet weight–dry weight) /wet weight × 100%.
Results were presented as mean ± SD. The data were assessed by unpaired tailed Student’s t test. P<0.05 was considered significant.
We observed upregulation of C1q expression in IBZ of pMCAO rats on day 5 after pMCAO compared to control rats without pMCAO(Figure 1A). We then evaluated effect of C1q on angiogenesis in the IBZ in pMCAO rats through silencing C1q expression by lateral ventricular injection of C1q siRNA. As illustrated in Figure 1B, suppression of C1q significantly reduced number of capillaries stained with FITC-anti CD31 antibody in the IBZ in pMCAO rats in comparison to control siRNA group, suggesting that C1q might be involved in angiogenesis following pMCAO in rats. Furthermore, we found that C1q levels were increased in RBMECs and HBMECs under hypoxia culture conditions (Figure 1C).
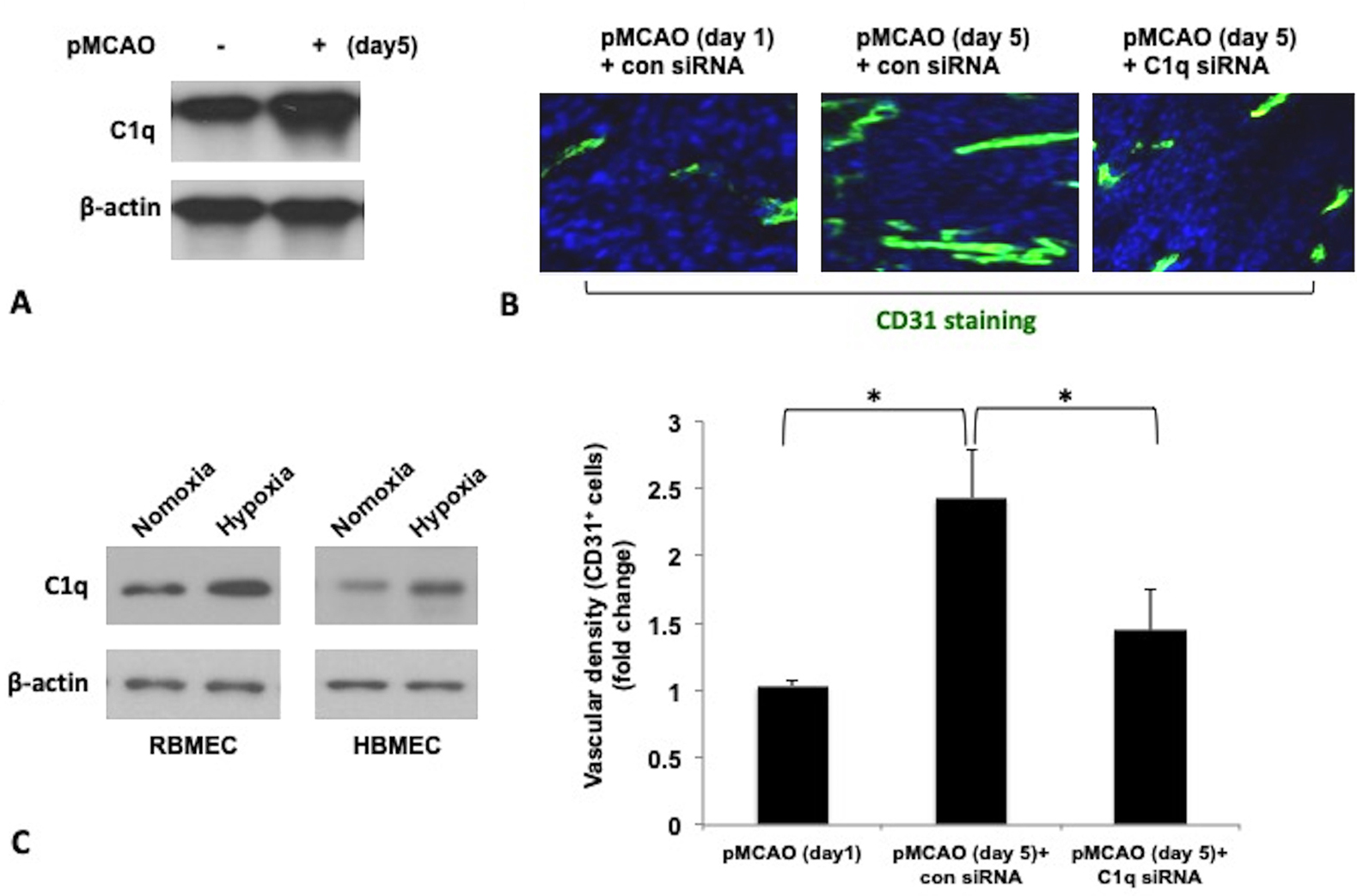 Figure 1.
Figure 1.
Expression of C1q in the IBZ in pMCAO rats, RBMECs and HBMECs under hypoxia conditions and its correlation with capillary density in pMCAO rats. (A) Western blot analysis of the expression of C1q in the IBZ of pMCAO rat on day 5 after pMCAO. (B) Vascular density in the IBZ in pMCAO rats was assessed using CD31 immunostaining (green) (scale bar =10 μm). Quantification of vascular density was shown as fold change in comparison to the pMCAO ( day 1) control group . (C) Expression of C1q in RBMECs and HBMECs under normoxia and hypoxia conditions. β-actin served as internal control. Data were presented as mean ±SD from three independent experiments. *P < 0.05.
We then explored whether C1q could regulate function of RBMECs and HBMECs. We examined the effects of C1q on viability, migration and tube formation of RBMECs and HBMECs. As shown in Figure 2A, viability of RBMECs and HBMECs was significantly increased by C1q (10 μg/ml). Transwell migration assay demonstrated that C1q treatment dramatically induced migration of both cells (Figure 2B). In addition, increased tube formation was observed in RBMECs and HBMECs treated with C1q in comparison to that of control cells with no C1q treatment (Figure 2C).
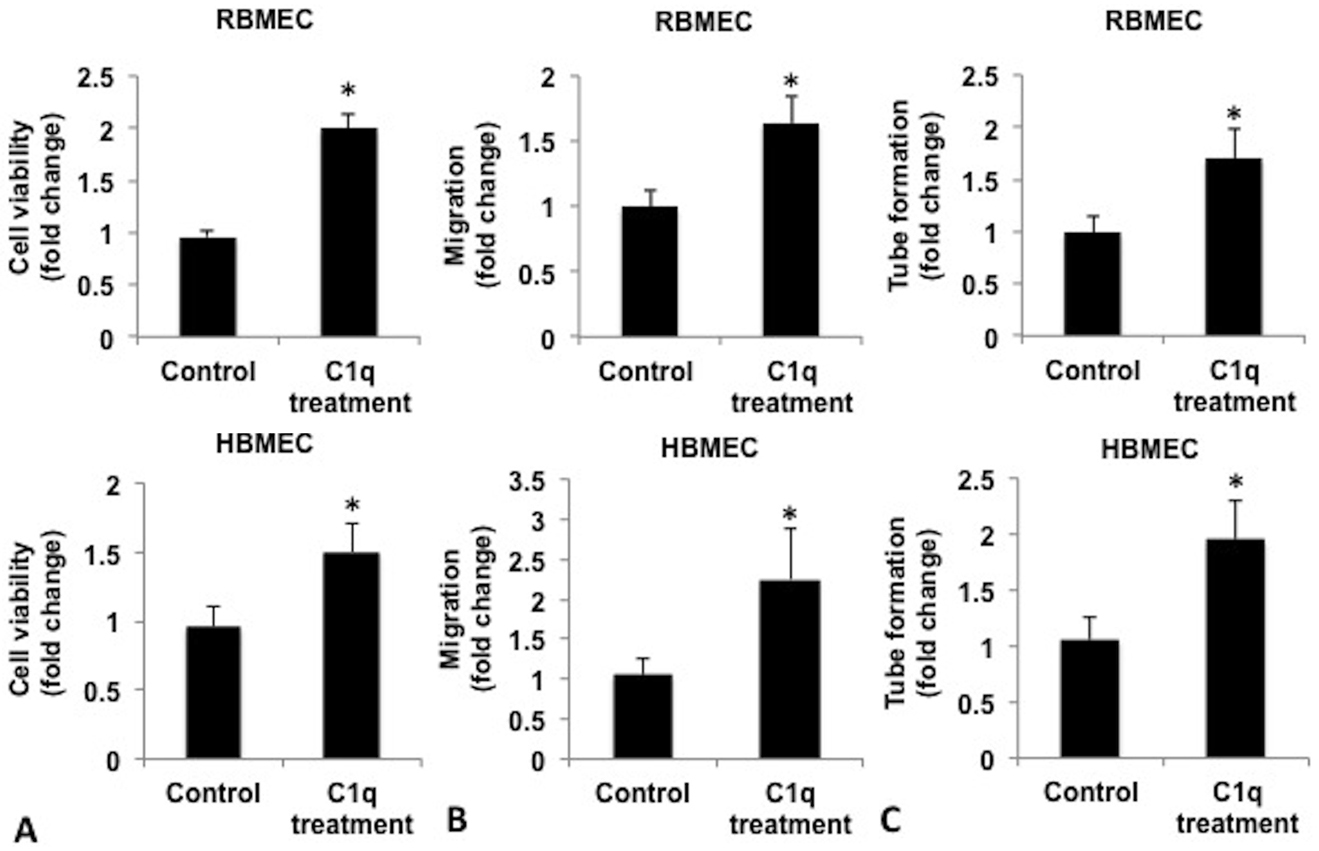 Figure 2.
Figure 2.
C1q enhanced angiogenic function of RBMECs and HBMECs. (A) Quantification of cell viability of RBMECs and HBMECs treated with C1q. RBMECs and HBMECs were cultured in the presence of 10 μg/ml C1q for 48 h. Cell viability was then measured by MTT assay. (B) Quantification of migration and tube formation (C) capacity of the cells was evaluated at 12 h and 24 h post C1q treatment respectively. For data analysis, the results were quantified as fold changes between control group and C1q treated group. Results were presented as mean ± SD from three independent experiments. *P<0.05 vs control group.
To further investigate the role of C1q in angiogenesis after pMCAO, we first examined VEGF expression and secretion in RBMECs and HBMECs. We found that VEGF secretion and expression were upregulated by C1q stimulation in the cells (Figure 3A). Also, enhancement in migration and tube formation in RBMECs and HBMECs by C1q treatment was abrogated by VEGF neutralizing antibody (Figure 3B and 3C). These findings suggest that upregulation of VEGF by C1q contributed to angiogenesis. Furthermore, increased VEGF expression was also observed in the IBZ in pMCAO rats and reduction in VEGF expression was correlated with C1q suppression by C1q siRNA in pMCAO rats (Figure 3D).
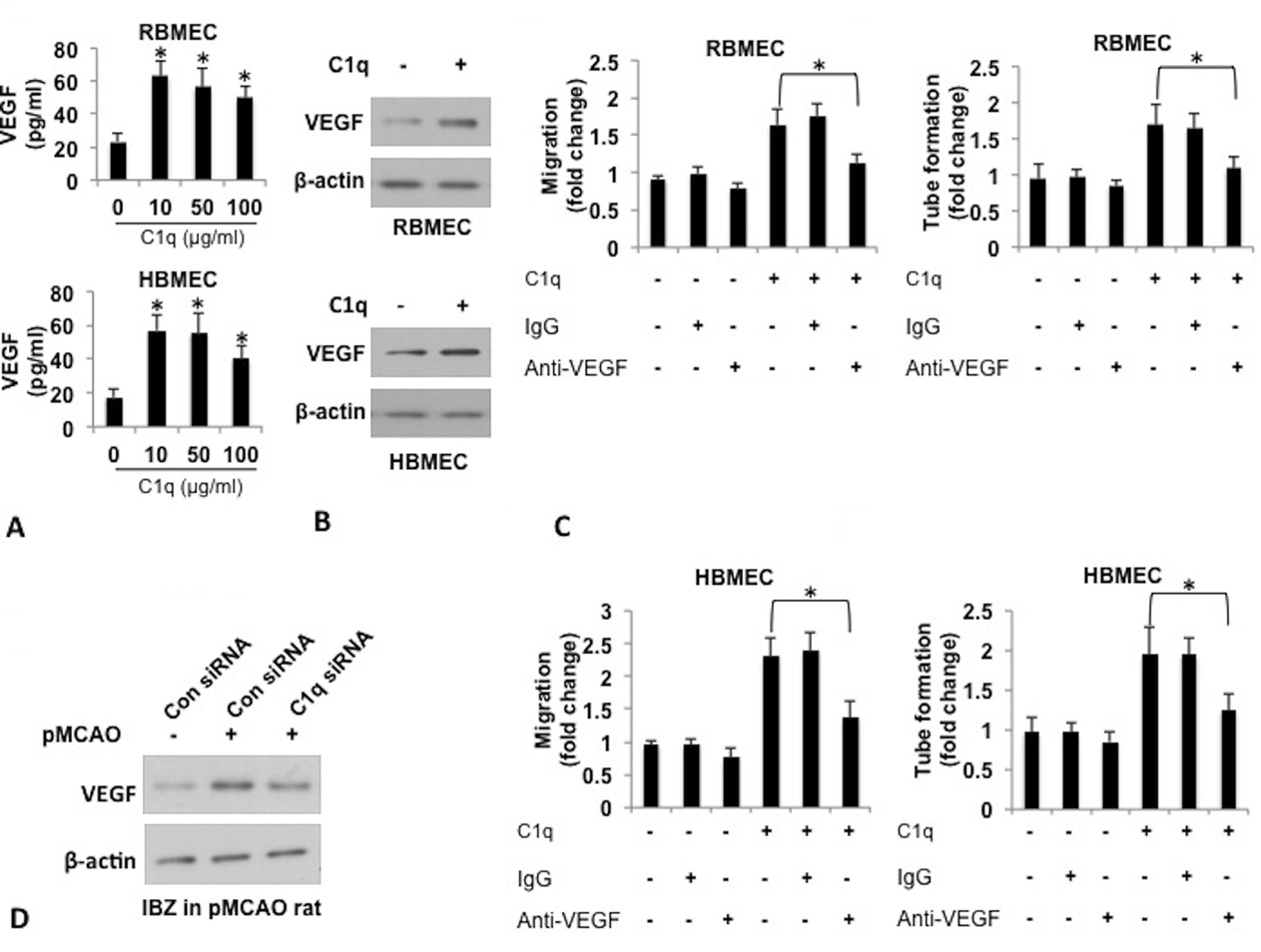 Figure 3.
Figure 3.
Regulation of VEGF secretion and expression by C1q. (A) Quantification of VEGF secretion in culture supernatant of RBMECs and HBMECs in the presence of various concentrations C1q and western blot analysis of VEGF expression in the cells. RBMECs and HBMECs were incubated with increasing concentrations of C1q (1, 10, 50, and 100 μg/ml) for 12 h. Levels of VEGF in culture supernatant were analyzed by ELISA. *P<0.05 vs control group with no C1q treatment. Expression of VEGF in the cells treated with 10 μg/ml C1q was examined by western blot analysis. β-actin served as internal control. (B) Quantification of migration and tube formation (C) capacity of RBMECs and HBMECs pretreated with VEGF neutralizing antibody in the presence of C1q. The cells were pretreated with 100 ng/ml VEGF neutralizing antibody for 1h, followed by C1q treatment. Migration and tube formation were evaluated at 12 and 24 h respectively post C1q treatment. Mouse IgG was used as IgG control to anti-VEGF antibody. For data analysis, the results were quantified as fold changes between control group with no treatment, and C1q, anti-VEGF or combination of C1q and anti-VEGF treated group. *P<0.05. (D) Western blot analysis of VEGF expression in the IBZ in pMCAO rats with or with no C1q suppression by siRNA. Results were presented as mean ± SD from three independent experiments. *P<0.05.
To investigate molecular mechanisms underlying enhanced angiogenesis after the stroke by C1q, we examined the role of C1q receptor LAIR1, in VEGF expression, migration and tube formation of RBMECs. As shown in Figure 4A, LAIR1 was expressed in both RBMECs and HBMECs. We also tested the expression of other C1q receptors, such as cC1qR, gC1qR, CR1, and LAIR2, and these receptors were undetectable in RBMECs and HBMECs (data not shown). LAIR1 expression in RBMECs was silenced by LAIR1 siRNA, and we found that silencing LAIR1 in RBMECs that were treated with C1q, significantly abrogated C1q-enhanced VEGF expression (Figure 4B), migration (Figure 4C) and tube formation (Figure 4D). It is interesting that levels of HIF1α, an upstream regulator of VEGF and VEGF were both reduced by LAIR1 inhibition and HIF1α siRNA, and the changes in expression of VEGF correlated with HIF1α levels (Figure 4E). These results suggest that enhancement of vascularization by C1q after the stroke might happen at least via LAIR1-HIF1α-VEGF pathway. Further studies need to be conducted to elucidate whether C1q could also directly regulate VEGF expression in a HIF1α independent fashion.
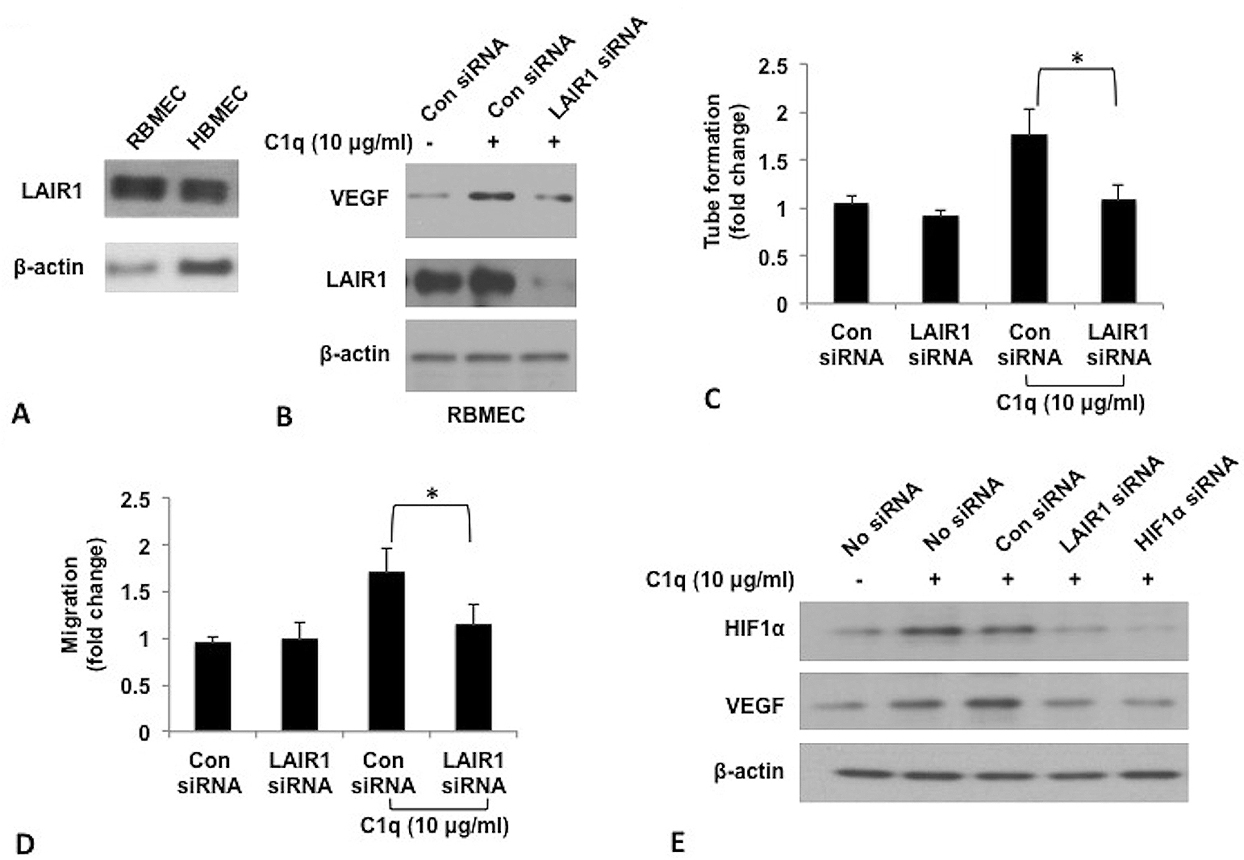 Figure 4.
Figure 4.
Role of LAIR1 and HIF1α in enhanced angiogenic function of RBMECs by C1q. (A) Western blot analysis of LAIR1 expression in RBMECs and HBMECs. (B) Western blot analysis of LAIR1 and VEGF expression in RBMECs with LAIR1 silencing by siRNA in the presence C1q. RBMECs transfected with control siRNA or LAIR1 siRNA were incubated with C1q (10 μg/ml) for 12 h. Expression of VEGF was then analyzed by using western blot. (C) Quantification of migration and tube formation capacity (D) of RBMECs treated as described above. (E) Western blot analysis of expression of HIF1α and VEGF in the RBMECs. For data analysis, the results were quantified as fold changes between control siRNA group without C1q treatment and C1q treated groups. Results were presented as mean ± SD from three independent experiments. *P<0.05 vs control group.
To evaluate the therapeutic effect of C1q in pMCAO rats, C1q was injected into lateral ventricles of the brain in pMCAO rats, and the capillary density in the IBZ was measured at day 5 post pMCAO. We found that C1q treatment significantly increased capillary density assessed by staining with FITC-anti CD31 antibody in the IBZ in comparison to human albumin treatment (Figure 5A). Also, pMCAO rats had greater edema in the ipsilateral (lpsi) hemisphere than normal rats and brain edema was dramatically reduced in pMCAO rat treated with C1q (Figure 5B).
 Figure 5.
Figure 5.
Therapeutic effect of C1q on vascularization in pMCAO rats. (A) Vascular density was assessed using CD31 immunostaining (green) (scale bar =10 μm). For data analysis, vascular density in the IBZ in pMCAO rats was quantified as fold changes between pMCAO with albumin treated group and pMCAO with administration of C1q group. *P < 0.05 vs albumin control group. (B) Changes in brain water content after C1q treatment. 5 days after pMCAO, cerebral edema was quantified as changes in brain water content in Ipsi hemisphere expressed as percentage of wet weight. n=5 per group. Results were presented as mean ± SD from three independent experiments. *p<0.05.
In this study, we investigated the ability of C1q and its molecular mechasnim to improve the recovery of stroke in pMCAO rats through enhanced neovascularization. We demonstrated that C1q expression was upregulated in the IBZ in rats with pMCAO and upregulation of C1q expression was associated with enhanced angiogenesis analyzed using CD31 immunostaining in the concerned area in the rats. Our in vitro and in vivo studies showed that treatment of C1q in RBMECs and HBMECs significantly promoted angiogenic functions of the cells through upregulating VEGF. Furthermore, molecular mechanisms underlying these findings are explored and we found that LAIR1, a receptor of C1q was required for regulation of angiogenic functions of RBMECs and angiogenesis in the IBZ in pMCAO rats by C1q. Moreover, HIF1α was an effector of C1q-LAIR1 signaling, which was involved in upregulation of VEGF.
It is known that compensatory angiogenesis is an important event during the post-stroke progression. Cells alter their gene profiles through transcriptional and post-transcriptional regulation to adapt to the new environment deficit in oxygen and energy (19). VEGF released by endothelial cells is the most potent proangiogenic factors involved in proliferation and migration of endothelial cells, increase in vascular permeability and degradation of the extracellular matrix, etc (20, 21). In our study, we found correlated expression levels of C1q and VEGF in the IBZ of pMCAO rats, RBMECs and HBMECs. Furthermore, proangiogenic function of C1q was abrogated by VEGF neutralizing antibody in HBMECs, suggesting that C1q exerts its vascular effect via VEGF instead of C1q itself. C1q may regulate VEGF expression by autocrine or paracrine mechanisms through its C1q receptor on endothelial cells.
We then sought to investigate the mechanisms underlying regulation of VEGF expression and angiogenesis by C1q following stroke. LAIR-1 is an inhibitory collagen receptor that inhibits cell signaling via the immunoreceptor tyrosine-based activation motif–containing collagen receptor complex. Although the function of LAIR-1 has been extensively studied in immune cells, its role in angiogenesis still remains uncertain (22). We demonstrated that LAIR-1 expressed in RBMECs was involved in C1q triggered upregulation of VEGF, migration and tube formation of the cells. These results suggest that an autocrine or paracrine regulation of neovascularization by C1q-LAIR-1 signaling might play an important role in post-stroke progression of vascularization. Interestingly, we also observed positive correlation between the expression of LAIR-1 and HIF1α, an upstream regulator of VEGF, in RBMECs in the presence of C1q.
HIF-1 is a heterodimeric basic helix-loop-helix protein formed by HIF1 α and β units, and activation of HIF1 that primarily occurs via HIF1α can induce expression of many hypoxia-inducible genes in hypoxic cells (23, 24). In vivo and in vitro studies have proved that VEGF expression is regulated by HIF1α at the transcriptional level. Higher levels of HIF1α can produce higher levels of VEGF through activation of DNA binding activity of HIF1α to VEGF promoter (25). In the present study, it is surprising that there was increased HIF1α expression in RBMECs treated with C1q and down-regulation of LAIR1 by siRNA reduced this HIF1α upregulation, suggesting that LAIR1 contributed to HIF1α expression and might act as an upstream regulator of HIF1α under normoxia conditions. We also examined the effect of C1q and LAIR1 silencing on HIF1β expression in the cells and found there were no changes in HIF1β levels (data not shown). HIF1α expression in the presence of C1q in RBMECs is likely controlled by either enhancement of expression or inhibition of degradation of HIF1α. Furthe studies are needed to investigate the mechanisms underlying the regulation of HIF1α expression by C1q-LAIR1 signaling and to confirm whether C1q-LAIR 1 could also directly modulate VEGF expression in HIF1α independent manner.
In conclusion, our study demonstrated that C1q upregulated VEGF at least via LAIR1-HIF1α-VEGF axis , and was thus involved in neovascularization after a stroke (Figure 6). Our findings suggest that C1q-LAIR1 signaling plays an important role in stroke recovery and may serve as a potential therapeutic strategy and improving prognosis following a stroke.
 Figure 6.
Figure 6.
Graphical demonstration on how C1q-LAIR1 operated to regulate angiogenic function of brain microvascular endothelial cells.
Dr. Guofeng Fan and Dr. Qiming Li contributed equally as co-first authors. The authors declare no conflicts of interest. We would like to thank ABM, Inc. for its English editing service.
Abbreviations: HBMECs: Human Brain Microvascular Endothelial Cells; RBMECs : Rat Brain Microvascular Endothelial Cells; HIF1α: Hypoxia Inducible Factor 1 α; IBZ: Ischemic Boundary Zone; LAIR1: Leukocyte-associated Inhibitory Receptor-1; pMCAO: Permanent Middle Cerebral Artery Occlusion; VEGF: Vascular Endothelial Growth Factor; PVDF: Polyvinylidene Difluoride