
A new method of coding of genetic information using laser speckles has been developed. Specific technique of transforming the nucleotide of gene into a speckle pattern (gene-based speckles or GB-speckles) is suggested. Reference speckle patterns of omp1 gene of typical wild strains of Chlamydia trachomatis of genovars D, E, F, G, and J are generated. This is the first report in which perspectives of the proposed technique in the bacterial gene identification and detection of natural genetic mutations in bacteria as a single nucleotide polymorphism (SNP) are demonstrated. The usage of GB-speckles can be viewed as the next step on the way to the era of digital biology.
As it is well-known, the nucleic acids (DNA or RNA) transmit and store the genetic information in living organisms. The structural and functional unit of hereditary information is the gene. A gene is a sequence of nucleotides in a molecule of nucleic acids. Typically, DNA consists of four types of nucleotides. These nucleotides contain, correspondingly, four nitrogen bases, namely, adenine (A), thymine (T), guanine (G) and cytosine (C).
The nucleotide sequence can be determined using the specific procedure of sequencing (1, 2) which allows presenting the primary structure of analyzing a macromolecule in the form of linear sequence of monomers in a text format.
All arbitrary sequences of nucleotides can be synthetized artificially, even if such sequence did not exist in nature before. This unique approach has been developed in pioneer investigations, which have been conducted recently by Microsoft Corporation (Microsoft Research). Papers (3, 4) are dedicated to the recording and storage of big numerical data using genetic methods and the development of DNA-based archival storage system. The above-mentioned studies devoted to the recording of digital information using synthetic strands of DNA are extremely important from the viewpoint of long-time storage of big data.
In the opinion of experts (5), we are currently facing a serious risk of data storage crisis. Thus, for example, the total amount of digital data generated in 2013 amounted to 3.5 zettabytes (Zb, 1 Zb = 1021 bytes), and 92 percent of these data in the world was generated only in 2012-2013. The exact information about the volume of data generated since 2014 up to present time has not been found in the open sources by the authors of this paper, but it is expected (5) the world will generate 40 Zb of data annually by 2020. It will not be possible, in principle, to provide the storage of such big data using traditional data media. However, as mentioned, such a problem can be easily resolved using data storage on DNA-carriers. Using synthetic DNA for the storage of numerical information makes it possible to achieve potential information density of 109 Gb/mm3 at the potential durability of thousands of years (6). Thus, the researchers of Microsoft Corporation (Microsoft Research) in co-operation with scientists from University of Washington have stored more than 200 Mb numerical data in the form of DNA (4, 6, 7, 8, 9). In particular, these data contain the encoding of high definition video (10), copies of the Universal Declaration of Human Rights in different languages, the top 100 books from Project Gutenberg, and the Crop Trust seed database (6). As it has been mentioned (9), the suggested algorithm of encoding a file containing digital data in DNA is robust to outliers and high levels of noise and is characterized by higher accuracy in comparison to similar methods.
Remarkably, those studies in the field of encoding numerical data on synthetic strands of DNA have also been performed by several other scientific teams, in parallel with Microsoft Corporation. Thus, in 2012, Harvard Medical School researchers encoded (11) in DNA a digital full-text book Regenesis: How Synthetic Biology Will Reinvent Nature and Ourselves and stored it on DNA-carrier (12). Later, in 2013, the European Bioinformatics Institute copied 739 kilobytes of sound, images, and text, including a 26-second audio clip of Martin Luther King, “I Have a Dream” speech. More recently, Harvard Medical School and a Technicolor research group reported storing and retrieving back 22 Mb of information that included a French silent film A Trip to the Moon (13).
However, the real essential progress has been achieved in very recent research published in Ref. (14) seria of images (five sequential frames from a digital movie) have been archived in the genome of living bacteria, keeping the ability of reproduction. It is also expedient to mention the papers (15, 16, 17, 18) directly referring to the problem of encoding numerical data using DNA carries of information.
So, at the moment, the main trend and the most perspective approach to the storage of big data is the usage of DNA-based carriers. However, at present time, genetic data themselves (including the big data) are sequenced and stored then atavistically in the text or numerical format. Definitely, the DNA sequencing is absolutely necessary for obtaining and pre-processing primary genetic information. However, further processing of very long nucleotide sequences is forestalling a serious problem in bioinformatics, especially when the critically fast growth of gene database is taken into consideration. The matter is that finding similar or identical fragments in nucleotide sequence of two different genes requires a lot of computations, especially in the case of large size genes. In order to find either highly similar or identical fragments in the nucleotide sequences of two different genes being compared, the sequences need to be analyzed in depth using several routine steps, which, however, require considerable time. In fact, it is necessary to conduct additional repeated sequencing/re-sequencing (sometimes up to 3-5 times) of the original sample with a repeated multi-stage computer analysis by several Genomic packages. This procedure is time-consuming (from a few days to several weeks), requires costly equipment, special facility and highly-qualified personnel. However, it is critical to avoid a significant amount (up to 20 percent) of false nucleotide polymorphisms at the sequencing stage when the primary nucleotide reads are being prepared. Moreover, in this case there is a certain difficulty related to interpreting the results obtained. Apparently, the algorithm used could produce a serious inconvenience and complicate significantly the study of the data even when small nucleotide sequences (250-500 bp in size) are analyzed. Working with nucleotide sequences of individual genes of a larger size (1000-1300 kb and more) is considered to be the greatest challenge despite the availability of a large number of different sequencing strategies for extended DNA fragments. Numerous errors of reading and analysis are overcome by repeatedly reading different fragments of the gene with the same package of multi-stage computer calculations. Working with such large nucleotide sequences obviously is 10-20 times more difficult and time-consuming than with the short ones (19).
But, if nucleotide sequence is recorded in analog format on diffraction optical element (DOE) or on hologram (HOE), then such artificial optical element can be used in the design of optical processor (20). In other words, the usage of speckles may be extremely useful in real-time optical processing of very long nucleotide sequences, development of express-methods of gene identification and typing, or in the holographic storage of genetic information.
Thus, the transformation of genetic data into either HOE/DOE or the presentation of nucleotide sequences in the form of a gene-based speckle structure will lead both to the significant improvement in the existing bioinformatics tools and to the creation of novel ones. This is critical for improving the methods of laboratory diagnostics of the infectious and non-infectious diseases of humans and animals.
Nevertheless, to the knowledge of the authors of this paper, at the present time, interdisciplinary research in the field of coherent optics and molecular biology has not been carried out. The authors of this paper tried to fill that gap. Earlier, a nucleotide sequence of omp1 gene of bacteria Chlamydia trachomatis (genovars D, E, F, G, J and K) has been successfully converted into 2D speckle-patterns. A special term, GB-speckles (gene-based speckles) has been introduced in Ref. (21, 22, 23) for the definition of a principally new class of speckles. Using GB-speckles as a new excellent tool for diagnostics is the next step to the era of digital biology. GB-speckles possess unique statistical properties, habitual for the speckles with a small number of scatterers (24, 25, 26) or so-called small-N speckles (27). The statistics of GB-speckles has been particularly investigated in Ref. (22). As it has been shown in Ref. (21), using such methods of speckle-optics, as speckle-correlometry, speckle-interferometry and subtraction of speckle-images allows defining the presence of natural mutations in comparing strains even in the case of a single SNP. It has been demonstrated in Ref. (21) that the appearance of any type of mutations leads to the formation of system of interferential fringes in the interference pattern in the case of using speckle-interferometric technique, which may serve as the basis for the operation of optical processor of genetic information. The optimization of the algorithm of encoding a nucleotide sequence of bacteria C. trachomatis into 2D GB-speckle pattern has been carried out in Ref. (23); it has been shown that the algorithm used in Ref. (21) is close to the optimal one.
The method of virtual phase-shifting speckle-interferometry (4-bucket technique) has been applied (28) to the investigations of polymorphism of two variants of omp1 gene of C. trachomatis (namely, strains E/Bour (E1 subtype) and E/IU-4 2 0755u4 (E2 subtype)).
This approach has already been successfully used for the detection of the C. trachomatis omp1 gene of the 11 known subtypes of this bacteria with genetic mutations in the form of either a single SNP or a combination of several SNPs, as previously reported by us, see Ref. (23). The nucleotide sequences of genes encoding the production of serine proteases, the Omptin family proteins of Enterobacteriaceae, which are known to be the causative agents of such infections as salmonellosis, yersiniosis, shigelosis and escherichiosis, have been successfully transformed into the format of GB-speckles. Such genes as pla (Yersinia pestis), pgtE (Salmonella enterica), sopA (Shigella flexneri), ompT and ompP (Escherichia coli), have been recently studied using the relevant GB-speckles (29).
The present paper is devoted to the study of fundamental principles of optical processing of GB-speckles for SNP detection in bacterial gene.
The methods of transformation of omp1 gene sequence in 2D speckle patterns are described in details. The effectiveness of the proposed technique for gene identification and detection of minimal differences between strains within the bacterial species is demonstrated. Comparative analysis of a six variants of omp1 gene from C. trachomatis six different genovars, which differ only in a single SNP(s), have been used to illustrate the process of computer generation of GB-speckles and the procedure of their processing.
At the first stage, the sequence of letters (taken from initial nucleotide sequence) is transformed in the sequence of numbers according to the following rule:
A -> 1, C -> 2, G -> 3, T -> 4.
It is important to note, that, as it has been found in Ref. (24) the functional relationship between numbers and letters is not so critical is this case. In other words, any other rule can be used for encoding, for example:
T -> 1, G -> 2, C -> 3, A -> 4.
Then all the possible associations (triads), containing only three numbers, which are combined from initial set of four numbers {1, 2, 3, 4}, are generated. These triads are:
(1 1 1), (1 1 2), (1 1 3), (1 1 4), (1 2 1), (1 2 2), (1 2 3), (1 2 4), (1 3 1), …, (4 4 4).
The total number of associations of four numbers, jointed in the triads, is equal to 64.
Then, at the next stage number 2, some discrete value h is assigned to each possible triad according to a very simple algorithm, described in Ref. (21). Value h is the integer, ranging from 1 to 64. Each triad from nuclear sequence corresponds to only one level of h. For example, association (1 1 1) corresponds to h=1, association (1 1 2) corresponds to h=2, (1 1 3) corresponds to h=3, (1 1 4) corresponds to h=4, (1 2 1) corresponds to h=5, (1 2 2) corresponds to h=6, etc. And finally, the last combination (4 4 4) corresponds to h=64.
At the step number 3 square matrix Hn,m is packed from vector h. Each element of this matrix relates to corresponding triad in the gene sequence. Typically nucleotide sequence of omp1 gene of C. trachomatis contains 394 triads. The size of square matrix H, closely matched with vector h (with the length of 394 elements) for C. trachomatis, is 20 x 20 elements. So, the last elements (since 395th element go 400th element) in vector h are filled by zeros before the packaging.
The physical meaning of formed matrix H is the local height of virtual scattering surface, reflecting the local content of gene structure. This surface will be used to generate unique speckle pattern, related to each specific nucleotide sequence of omp1 gene of C. trachomatis.
The physical meaning of the formed matrix H consists in the fact that each element of this matrix represents by itself the local height of some virtual rough surface, related to the local content of analyzing genetic structure. The obtained virtual rough surfaces will be used for simulation of unique speckle-patterns, conforming to different specific nucleotide sequences.
2D speckle pattern, corresponding to initial nucleotide sequence, is generated using the diffraction of coherent beam with square profile on the (virtual) rough surface with profile Hn,m. In each point of virtual diffuser, phase modulation Un,m=exp(-2pi iHn,m/64) is introduced. Surface is illuminated at norm by coherent beam with square profile; the phase in illuminating beam is a constant, the resolution of speckle-image is 2048 pixels x 2048 pixels. In this paper, computer simulation of process of formation of GB-speckles has been carried out.
The example of re-coding of omp1 gene of C. trachomatis, genovar D, strain B-120, subtype D1 is shown in Figure 1.
 Figure 1
Figure 1The example of re-coding of omp1 gene of C. trachomatis.
The surface plot (it is important to note, that in this paper the intensities in all pictures are re-normalized into the range [0; 255] grey-scale levels) of generated matrix Hn,m, corresponding to the omp1 gene of C. trachomatis strain B-120, is shown in Figure 2.
 Figure 2
Figure 2Surface plot of generated matrix Hn,m of the omp1 gene of C. trachomatis strain B-120.
The whole algorithm is shown schematically in Figure 3. Speckle pattern (spatial intensity fluctuations) is obtained after performing the 2D fast Fourier transform of Un,m. Gene-based speckle pattern corresponding to the structure of the omp1 gene C. trachomatis strain B-120, subtype D1 is shown in Figure 4.
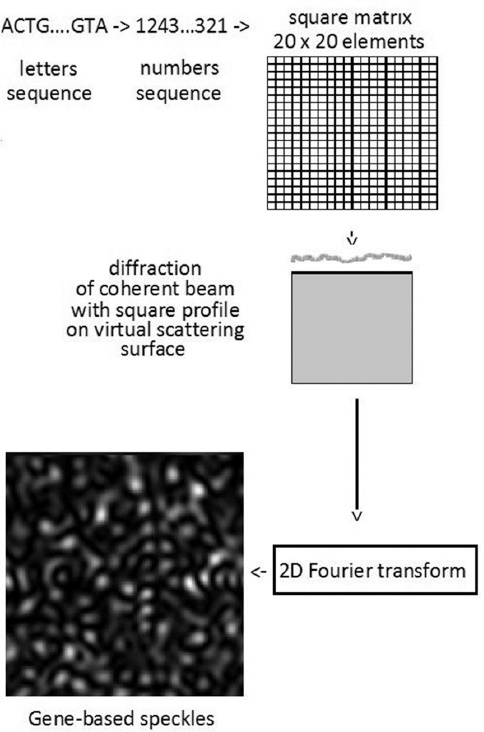 Figure 3
Figure 3Illustration of the algorithm of transforming the nucleotides sequence of the model micro-organism studied (the omp1 gene of C. trachomatis strain B-120) into a gene-based speckle pattern.
 Figure 4
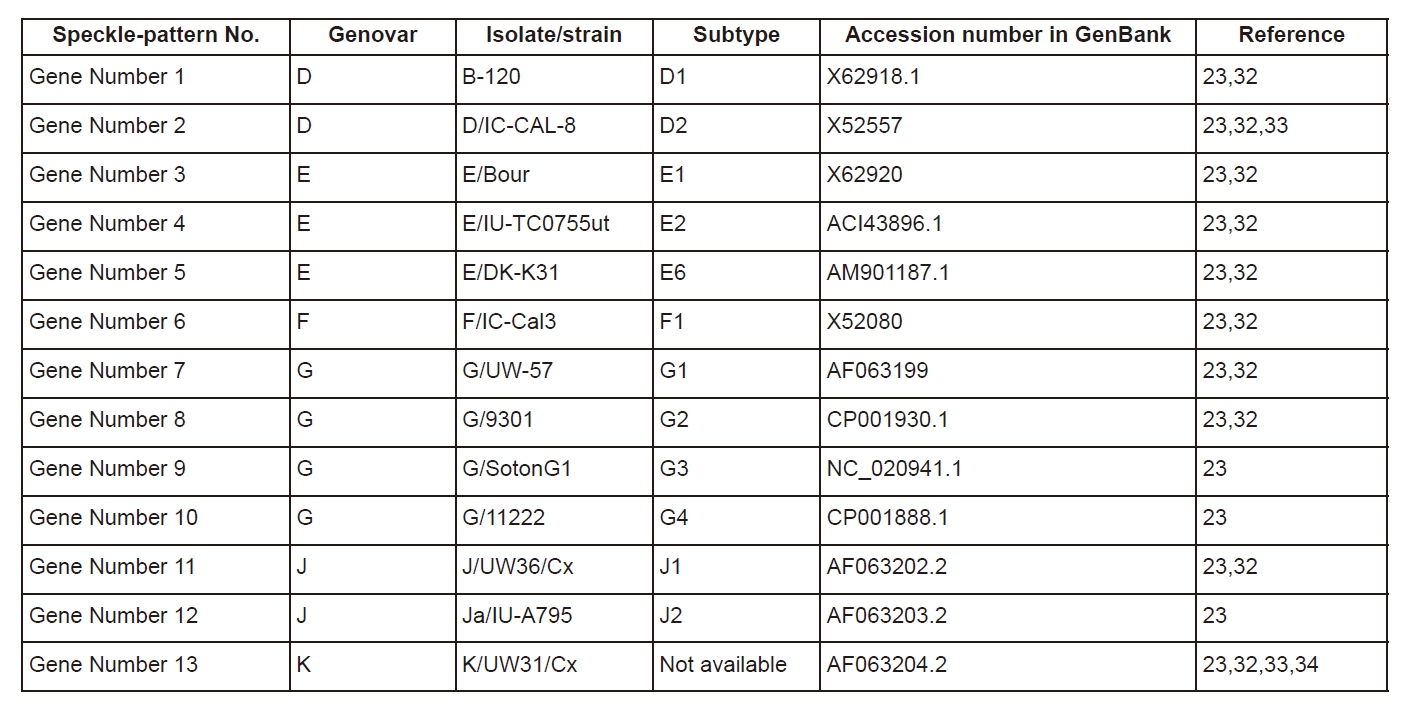
Figure 4Examples of speckle patterns, corresponding to the omp1 gene of C. trachomatis of different genovars and subtypes (see Table 1). A. Gene number 1. B. Gene number 2. C. Gene number 3. D. Gene number 4. E. Gene number 5. F. Gene number 6. G. Gene number 7. H. Gene number 8. I. Gene number 9. J. Gene number 10. K Gene number 11. L. Gene number 12. M. Gene number 13.
When speckle patterns corresponding to different genes are simulated, they then can be compared using the methods of speckle-metrology (30, 31). In other words, similarities and differences between compared nucleotide sequences can be found using the interference of speckle-patterns, speckle subtraction and cross-correlation techniques.
Using the algorithm described in Section 2.1 and Section 2.2 speckle patterns for omp1 gene corresponding to thirteen different strains of six genovars of C. trachomatis have been generated. The following C. trachomatis omp1 gene nucleotide sequences used are presented at Table 1.
It is expedient to note that, overall, thirteen compared genes of C. trachomatis are characterized by high level of identity. That is why the differences between corresponding speckle patterns are also negligible (for example, compare speckle-pattern, constructed for gene number 1 and speckle-pattern, obtained for gene number 2, in Figure 4). Nevertheless, even small differences are easily detectable in the GB-speckles. If differences between the compared omp1 gene sequences are essential, the differences in corresponding speckles are also higher (compare, for example, number 4, number 6, number 13 in Figure 4). Clearly, if the similarity between the compared gene sequences is higher, then the value of correlation coefficient of speckles is also higher.
Cross-correlation coefficients of speckle patterns are presented in Table 2. Evidently, those diagonal elements of matrix present themselves as the coefficients of auto correlation, consequently they always equal unity. The matrix is symmetrical because the property of correlation corr (A,B)=corr (B,A) for all square matrices. The absolute value of cross-correlation coefficient varies within the range [0.006; 0.998], which reflects the degree of similarities of compared nucleotide sequences.
As it has already been noticed, if we compare the same omp1 gene of C. trachomatis strains within a single genovar of different subtypes, they seem to be very similar. The differences in corresponding speckle patterns are minimal, but detectable. One way to enhance specific features in analyzed patterns is to use speckle subtraction. As it can be clearly seen from Figure 5A, a lot of new minutia, characterizing speckle differences, are developed after the application of speckle subtraction technique.
 Figure 5
Figure 5Processing of the speckle pattern, based on gene number 1 and the speckle pattern, based on the gene number 2. A. Speckles subtraction. B. Interference of the speckles.
However, the difference between the interference picture shown in Figure 5B and the initial speckle pattern shown in Figure 4B is practically negligible. In the case when pi phase shift is introduced, but interfering speckles are not identical, structural changes in interference picture occur (see Figure 6A). Instead of random speckles, quasi-regular interference fringes develop in the resulting interference pattern. In the case when speckle patterns based on genes with essential differences interfere, large size fringes modulated by small speckles (see Figure 6B) may show up. Sometimes, when a gene sequence contains identical, but shifted fragments, the internal structure of fringes acquires very specific features, see Figure 6C-E.
 Figure 6
Figure 6Interference of the two speckle-patterns. Constant pi phase shift between speckle-patterns is introduced. A. different speckles (pattern generated on the base of gene number 1 interferes with pattern based on gene number 2 with pi phase shift). B. Different speckles (pattern based on gene number 1 interferes with pattern based on gene number 7 with pi phase shift). C. Different speckles (pattern based on gene number 7 interferes with pattern based on gene number 9 with pi phase shift). D. Different speckles (pattern based on gene number 7 interferes with pattern based on gene number 10 with pi phase shift). E. Different speckles (pattern based on gene number 7 interferes with pattern based on gene number 11 with pi phase shift).
It is clearly seen, that if identical gene-based speckles interfere, then the resulting interference picture does not contain any fringes. However, high-contrast fringes appear in the case of interference of non-identical gene-based speckles, even in the case if differences in compared genes are negligible. This means that speckle-interferometry is a powerful technique of gene subtyping, at least for the bacterial model used (C. trachomatis).
There are several main types of well-known genetic bacterial mutations: deletion, insertion, substitution of either a single or several nucleotide(s) in a single gene. In computer simulation gene number 1 has been selected as an example of an intact gene and three more sequences with mutations have been imitated. In the first simulated sequence, one nucleotide, selected at random at the beginning of the initial nucleotide sequence has been deleted. In the second sequence, the positions of two neighboring nucleotides have been reordered (also at the beginning of initial sequence). And finally, one more nucleotide has been added at the beginning of initial sequence for the purpose of simulating the mutation of insertion type.
The speckle-patterns related to the mutations described above are shown in Figure 7. In the case of deletion (see Figure 7A), speckles are only shifted in comparison with reference speckle-pattern (compare with Figure 4A), without essential decorrelation. In the case of a substitution of nucleotide (see Figure 6B), a practically total decorrelation of the observed speckle pattern occurs. If mutation of an insertion type is generated, then the decorrelation of the speckle pattern is very weak (compare Figure 4A and Figure 7C). So, all types of mutations can be detected using speckle-correlometry, but the evident recognition of mutations is guaranteed only in the case of insertions. At the same time, it is critically important to emphasize that speckle-interferometric technique is very sensitive to any type of genetic mutations. For example, the speckle pattern forming as a result of interference of reference speckle pattern shifted on pi by phase with speckles related to mutation by type of deletion is shown in Figure 7D. It is clearly seen that initial speckle pattern is structurally changed: instead of random speckles, parallel horizontal fringes can be observed.
 Figure 7
Figure 7Detection of single mutations in the omp1 gene of C. trachomatis strain B-120 of genovar D. A. Deletion. B. Insertion. C. Substitution. D. Interference pattern, relating to gene deletion.
Here, it is shown that the methods of speckle-correlometry and speckle-interferometry can be efficiently used for the detection and interpretation of genetic information. Based on computer simulation it has been demonstrated that an optical processor for SNP detection exploring GB-speckles can be designed. Even a small difference in compared nucleotides sequences leads to essential changes in corresponding computer generated speckle patterns.
The interference of GB-speckles based on nucleotide sequence is very sensitive to any type of mutations. The recognition of fringes appearing in interference pattern may serve as the basis for the express-method of detecting genetic mutation and opens the way to designing an optical processor for identifying the gene and typing both pathogenic and non-pathogenic bacteria. Speckle-interferometry of gene-based speckles is a very perspective technique for the study of Chlamydia diversity in particular, as well as bacterial diversity in general.
This study has been supported by the Russian Science Foundation, Grant No. 17-16-01099.
Abbreviations: bp, base pairs; C. trachomatis, Chlamydia trachomatis; DNA, deoxyribonucleic acid; DOE, diffraction optical element; Gb, gigabyte; GB-speckles, gene-based speckles; HOE, holographic optical element; Kb, kilobyte; omp1, gene encoded the outer membrane protein 1 in C. trachomatis; ompP, gene encoded the outer membrane protease OmpP in Escherichia coli; ompT, gene encoded the outer membrane protease OmpT in Escherichia coli; pgtE, gene encoded the PgtE protease in Salmonella enterica; Pla, Plasmin activator; RNA, ribonucleic acid; SNP, single nucleotide polymorphism; sopA, gene encoded the SopA protease in Shigella flexneri; Zb, zettabyte.