







Frontiers in Bioscience-Landmark (FBL) is published by IMR Press from Volume 26 Issue 5 (2021). Previous articles were published by another publisher on a subscription basis, and they are hosted by IMR Press on imrpress.com as a courtesy and upon agreement with Frontiers in Bioscience.

1 Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Flemingovo n. 2, Prague 6, 16610, Czech Republic
2 First Faculty of Medicine, Charles University, Katerinska 32, Prague 2, 12108, Czech Republic
3 Department of Biochemistry, Faculty of Science, Charles University, Albertov 6, Prague 2, 12843, Czech Republic
Abstract
Glutamate carboxypeptidases II and III (GCPII and GCPIII) are highly homologous di-zinc metallopeptidases belonging to the M28 family. These enzymes are expressed in a variety of tissues, including the brain, prostate, kidney, testis and jejunum. GCPII has been recognized as a neuropeptidase in the central nervous system, as a folate hydrolase participating in absorption of folates in the jejunum and, most importantly, as a prostate-specific membrane antigen that is highly expressed in prostate adenocarcinoma. Furthermore, it has been identified in the neovasculature of most human solid tumors. In contrast, GCPIII has not been associated with any specific physiological function or pathology, and its expression, activity and inhibition have not been as well-studied. In this review, we provide an overview of the current understanding of the structure, enzymatic activity, substrate specificity, and tissue distribution of these two homologous enzymes. We discuss their potential physiological functions and describe the available animal models, including genetically modified mice. We also review the potential use of specific monoclonal antibodies and small-molecule inhibitors recognizing GCPII/III for diagnosis, imaging and experimental therapy of human cancers and other pathologies.
Keywords
- GCPII
- GCPIII
- PSMA
- FOLH1
- NAALAD2
- NAAG
- Folate
- BCG
- Review
Metallopeptidases require bivalent metallic cations for catalysis. According to the MEROPS database (1), metallopeptidases can be divided into 99 peptidase families based on statistically significant similarities in their amino acid sequences. Each family is typically characterized by additional features. For example, the M28 peptidase family contains aminopeptidases and carboxypeptidases that require co-catalytic zinc for their enzymatic activity. This family consists of six subfamilies, with subfamily M28B containing glutamate carboxypeptidase II (GCPII) and glutamate carboxypeptidase III (GCPIII).
GCPII and GCPIII are type II transmembrane glycoproteins with a short intracellular portion, a single membrane-spanning segment, and a large extracellular domain with carboxypeptidase activity (Figure 1) (2-4). In the prostate, GCPII is referred to as prostate-specific membrane antigen (PSMA), and is highly expressed in prostate adenocarcinoma (5). It also has been identified in the neovasculature of most solid tumors (6). GCPII has been independently described in the brain as N-acetylated alpha-linked acidic dipeptidase (NAALADase) and in jejunum as a folate hydrolase responsible for absorption of folates (3, 7-9). While the physiological function of GCPII has been well-characterized in the nervous system and small intestine, it is still not fully understood in other highly GCPII-expressing tissues such as the prostate and kidney. In contrast to the wealth of information about GCPII, only a few reports describing GCPIII have been published to date. Its physiological function has yet to be fully elucidated, and there is no information available on its possible involvement in human diseases.
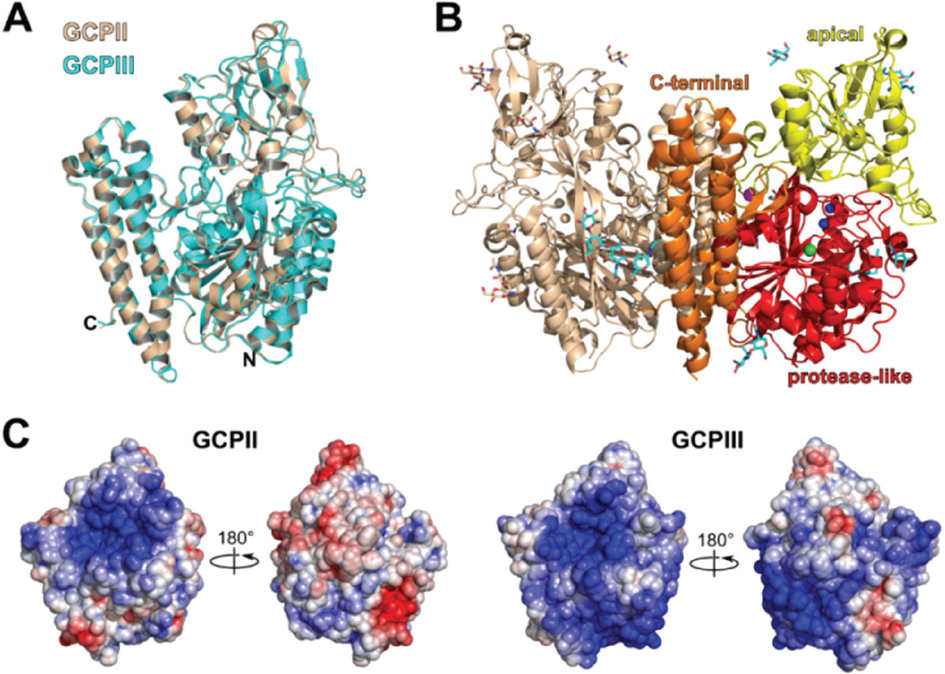 Figure 1
Figure 1Comparison of the overall 3D structures of GCPII and GCPIII. A: Structural alignment of GCPII (PDB 2PVW) and GCPIII (PDB 3FF3) monomers. Both monomers are depicted in cartoon representation. The GCPIII monomer is colored cyan, and the GCPII monomer is colored wheat. C- and N-termini of GCPII monomer are indicated. B: Crystal structure of GCPII homodimer in cartoon representation. One monomeric unit is shown in wheat while the other is colored according to the domain organization: the protease-like domain in red, the apical domain in yellow, and the C-terminal domain in orange. The zinc cations are shown as blue spheres, the chloride anion as a green sphere, and the calcium cation as a violet sphere. The carbohydrate moieties are shown as sticks in cyan. C: Comparison of the surface electrostatic potentials of the GCPII (PDB 2PVW) and GCPIII (PDB 3FF3) X-ray structures. Two representations of both structures are shown rotated by 180° (the left orientation being identical to the orientation of the molecule in panel A). The electrostatic gradient is shown from the most negatively charged surface (red) to the most positively charged surface (blue). The figures were created using The PyMOL Molecular Graphics System, Version 0.99 Schrödinger, LLC. The electrostatic potential maps were calculated using PyMol APBS Tools.
The genes encoding GCPII and GCPIII map to chromosome 11, and their exon/intron structures are similar (10). While the FOLH1 gene encoding GCPII is localized at position 11p11-p12 (2, 11, 12), the NAALAD2 gene encoding GCPIII is at position 11q14.3.-q21 (13). GCPII and GCPIII share 67% amino acid sequence identity and 81% sequence similarity. The overall 3D structures of these proteins are very similar, with a root-mean-square deviation (RMSD) for 676 aligned C-alpha atoms of 0.59 Å (see Figure 1A) (13, 14). The extracellular parts of both proteins form homodimers in which each monomer folds into three domains—the protease-like domain, the apical domain and the C-terminal or dimerization domain (see Figure 1B) (14-17). Both proteins are post-translationally N-glycosylated, which is crucial for their enzymatic activities (18-21).
Despite these many similarities, several structural differences between GCPII and GCPIII have been identified. These include differences in surface charge distribution that could be a major determinant in physiologically relevant interactions with hypothetical protein partners or ligands (see Figure 1C) and slightly divergent active sites that result in distinct substrate specificities and catalytic efficiencies (14).
This review aims to compare the enzymatic activities and expression profiles of GCPII and GCPIII under physiological and pathophysiological conditions. We describe known functions of GCPII in the brain and small intestine and discuss the potential roles of GCPII and GCPIII in these and other tissues. Finally, we provide an overview of the use of GCPII as a diagnostic and therapeutic target, describe the current state of inhibitor development, and discuss why it is of high interest to identify small-molecule compounds that selectively bind GCPII but not GCPIII.
As their names suggest, both GCPII and GCPIII are hydrolases capable of cleaving a C-terminal glutamate from their substrates. Indeed, both enzymes can process most N-acetylated dipeptides of the form Ac-X-Glu-OH, where X is any proteinogenic amino acid (21-23). The catalytic efficiencies of GCPII and GCPIII differ significantly for some dipeptides (21), which is not surprising because there are several important structural differences in the active sites of these enzymes.
First, while the occupancy of zinc ions within the GCPII active site reaches 100%, zinc ions within the GCPIII active site display much lower occupancies (80-95% for Zn1 and 40-80% for Zn2) (see Figure 2A) (14, 16). This is likely caused by substitution of Asn519 in GCPII with Ser509 in GCPIII. Ser509 has two different conformations in the GCPIII structure. In the first conformation, Ser509 interacts with the Zn2-coordinating amino acid Asp443 in a similar matter as Asn519 interacts with Asp543 in GCPII. However, in the second conformation, Ser509 does not interact with Asp443, enabling Asp443 to adopt an alternative conformation in which it does not coordinate the Zn2 ion. This conformational flexibility is likely the reason for the low occupancy of Zn2 within the GCPIII active site (see Figure 2A) (14, 24).
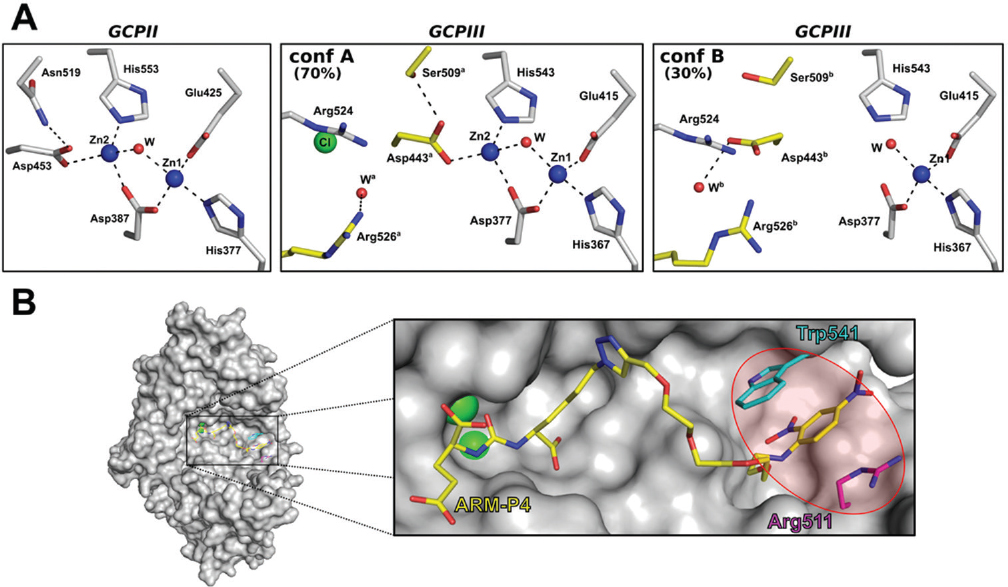 Figure 2
Figure 2Structural differences between the GCPII and GCPIII active sites. A: Comparison of the zinc coordination sites of GCPII (PDB 2OR4) and GCPIII (PDB 3FF3). For GCPIII, two different conformational states (A and B) are shown. The amino acids that adopt different conformations in the GCPIII structure are colored yellow, while the rest of the amino acids are colored gray. All amino acids are shown in stick representation. Oxygen atoms are colored red, and nitrogen atoms blue. Zinc ions are shown as blue spheres and the chloride ion as a green sphere. Water molecules are shown as red spheres. The hydrogen bonds and zinc co-ordinations are shown as black dashed lines. B: Highlight of the arene-binding site (ABS) within GCPII (PDB 2XEG). GCPII is shown in surface representation colored gray with a portion of the molecule omitted for visual clarity. The ligand ARM-P4 is shown as sticks and colored yellow. Trp541 and Arg511, the amino acids comprising the ABS, are shown as sticks and colored cyan and magenta, respectively. Oxygen atoms are colored red, and nitrogen atoms blue. Zinc ions are shown as green spheres. The figures were created using The PyMOL Molecular Graphics System, Version 0.9.9 Schrödinger, LLC.
Second, GCPII contains a so-called arene-binding site (ABS) that is absent in GCPIII (24-26). This exosite is formed by the Trp541 and Arg511 side chains at the entrance of a tunnel leading into the GCPII active site (see Figure 2B) and seems to be important for both processing of some endogenous substrates and binding of highly specific inhibitors (25, 26). Indeed, pi-pi stacking interactions with the Trp541 and Arg511 side chains participate in the binding of arene-containing ligands into the GCPII active site. In GCPIII, Trp541 is substituted by Lys531, which effectively prevents binding of similar ligands to the enzyme, as the Lys side chain is unable to engage in pi-pi stacking interactions.
To date, two physiologically relevant substrates processed by both GCPII and GCPIII have been identified—N-acetyl-L-aspartyl-L-glutamate (NAAG) and polyglutamylated folates (FolGlun) (13, 24). In addition, both enzymes have been shown to cleave naturally occurring β-citryl-L-glutamate (BCG), although the catalytic efficiency of GCPII is extremely low, thus making BCG a specific substrate of GCPIII (24).
Both GCPII and GCPIII cleave NAAG, yielding N-acetyl-L-aspartate and L-glutamate (Figure 3). NAAG hydrolyzing activity was first detected in 1984 on membranes isolated from rat brain cortex (27). Soon after, this activity was assigned to a membrane-bound metallopeptidase termed N-acetylated alpha-linked acidic dipeptidase (NAALADase) (28, 29), which was later designated as GCPII (8, 9, 30). In 1999, a second membrane-bound enzyme capable of NAAG hydrolysis was identified (13). This enzyme was first named NAALADase II but later designated as GCPIII (4). Since then, several studies comparing the catalytic efficiencies of GCPII and GCPIII have been published (4, 20, 21, 24).
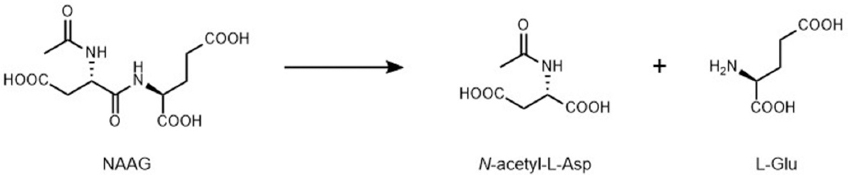 Figure 3
Figure 3NAAG-hydrolyzing reaction catalyzed by GCPII or GCPIII.
Bzdega et al. reported that rat GCPII and mouse GCPIII have similar catalytic efficiencies and pH dependencies for NAAG hydrolysis (4). In contrast, a comparative analysis of human GCPII and GCPIII performed by Hlouchova et al. using purified recombinant proteins revealed that GCPII processes NAAG with 3- to greater than 10-fold higher catalytic efficiency than GCPIII, depending on the reaction conditions. Moreover, the pH and salt concentration optima of human GCPII and GCPIII differ significantly (21).
Interestingly, GCPII and GCPIII respond differently to the presence of divalent metal ions during the NAAG-hydrolyzing reaction. While GCPII is essentially metal-insensitive, GCPIII NAAG-hydrolyzing activity can be stimulated by addition of Mn2+ or Zn2+ and inhibited by addition of Ca2+ (20, 24). This difference relates to replacement of Asn519 in GCPII with Ser509 in GCPIII. Recombinant GCPII becomes metal-sensitive when a N519S mutation is introduced (20). Because the occupancy of the Zn2 ion within the GCPIII active site is low, Zn2 in the GCPIII structure can be replaced with a different metal ion, creating a heterometallic cluster and altering the enzyme’s catalytic efficiency. Notably, a reciprocal mutation in GCPIII (i.e., S509N) does not abolish the metal dependency of the enzyme for NAAG-hydrolyzing activity, suggesting that the mechanism of metal ion involvement in the reaction is likely more complex (20).
GCPII was recently shown to process the endogenous tripeptide N-acetyl-L-aspartyl-L-glutamyl-L-glutamate (NAAG2) (31). Using recombinant enzymes, we demonstrated that not only GCPII but also GCPIII is capable of NAAG2 cleavage (Navratil et al., unpublished observation). However, the enzyme kinetics for this reaction has not yet been evaluated in detail.
The hydrolysis of polyglutamylated folates (FolGlun) by both GCPII and GCPIII yields folate and free L-glutamate (Figure 4) (24, 26). The role of GCPII as an intestinal folate hydrolase was identified by two independent research groups in the 1990s (7, 32), although detection of FolGlun hydrolyzing activity in intestinal mucosa dates back to 1969 (33). Our group recently reported that GCPIII also is able to cleave FolGlun, and we directly compared GCPII and GCPIII catalytic efficiencies in FolGlun-hydrolyzing reactions (24).
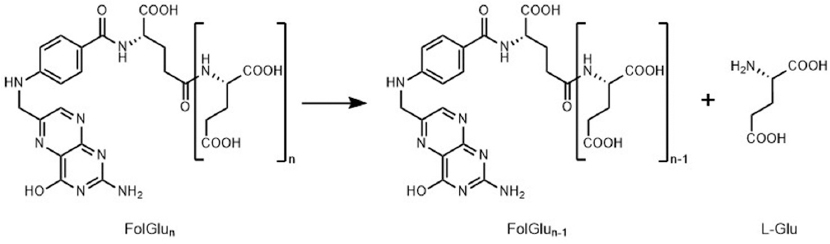 Figure 4
Figure 4FolGlun-hydrolyzing reaction catalyzed by GCPII or GCPIII.
Interestingly, while cleavage of monoglutamylated folate (FolGlu1) by the two enzymes is comparable, the processing of polyglutamylated folates (FolGlu2-6) by GCPII is almost two orders of magnitude more efficient than by GCPIII (24, 26). This observation can be explained by the engagement of the GCPII ABS in recognizing polyglutamylated folates. The ABS binds the hydrophobic pteroate moiety, leading to potential stabilization of FolGlu2-6 in the active site. As the ABS is absent in GCPIII, FolGlu2-6 cannot be stabilized in the GCPIII active site in a similar manner. When processing FolGlu1, GCPII is unable to fully utilize the ABS for binding of the pteroate moiety of the substrate, leading to cleavage kinetics comparable to that of GCPIII.
The metal dependence of GCPII and GCPIII catalytic efficiency has only been explored for FolGlu1 cleavage, and researchers found a similar trend as for NAAG cleavage. GCPII appears insensitive to metals during FolGlu1 processing. On the other hand, GCPIII shows the highest catalytic efficiency in the presence of Zn2+, followed by Mn2+ and Ca2+ (24).
Both GCPII and GCPIII cleave BCG, yielding citrate and L-glutamate (Figure 5) (24). BCG-hydrolyzing activity was first detected in the rat testis in 1983 (34), followed by purification of a BCG-hydrolyzing enzyme from the rat testis particulate in 1995 (35). However, identification of this BCG-hydrolase as GCPIII was published more than 15 years later (20). In their pivotal work, Collard and co-workers discovered the BCG-hydrolyzing activity of mouse GCPIII and reported that mouse GCPII is not capable of BCG cleavage. Using highly purified recombinant enzymes, we recently found that human GCPII can process BCG, but with catalytic efficiency three to five orders of magnitude lower than that of human GCPIII (24). These observations suggest that BCG can indeed be considered a specific substrate of GCPIII.
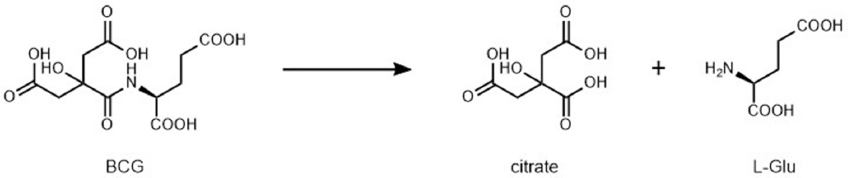 Figure 5
Figure 5BCG-hydrolyzing reaction catalyzed by GCPII or GCPIII.
The reasons for such a striking difference in the ability of these enzymes to process BCG are not fully understood, but they seem to be related to replacement of Asn519 in GCPII with Ser509 in GCPIII. Indeed, Collard and co-workers showed that introducing a N519S mutation into mouse GCPII renders the enzyme capable of BCG hydrolysis. Introducing the reciprocal S509N mutation in mouse GCPIII abolishes its BCG-hydrolyzing activity (20). The main reason that substitution of Asn519 in GCPII with Ser509 in GCPIII affects BCG hydrolysis is likely connected with the fact that this substitution influences the metal ion occupancy in the active site.
Similar to the NAAG-hydrolyzing and FolGlun-hydrolyzing reactions, BCG hydrolysis by GCPIII is stimulated by addition of certain divalent metal ions, while GCPII is metal-insensitive. Interestingly, the metal-dependency of GCPIII for BCG hydrolysis shows an almost completely opposite trend than in the case of NAAG cleavage—it is stimulated by addition of Mn2+ or Ca2+ and inhibited by addition of Zn2+ (20, 24). Collard and co-workers proposed that the presence of Mn2+ or Ca2+ in the reaction leads to generation of heterometallic Zn-Mn or Zn-Ca clusters within the GCPIII active site. Because BCG brings more negative charges into the active site than NAAG, metal ions with the ability to bind more ligands (6 or 7 ligands for Ca2+ compared with 4 or 5 for Zn2+) are preferable (20). However, QM/MM calculations by Navratil et al. did not confirm that Ca2+ would be thermodynamically more favorable for BCG as a substrate; this suggests that a more complex mechanism of action underlies the metal-dependency of GCPIII enzymatic activity (24).
Mentioned discrepancies could be addressed by solving the GCPIII structure in complex with BCG and appropriate metal ions. The comparison of such a structure with the available structure of the GCPII-BCG complex, together with QM/MM/MD calculations, could address not only why BCG is a specific substrate of GCPIII but also how metal ions are involved in the reaction.
Since the development of the first anti-GCPII antibody, 7E11 (5), several research groups have thoroughly studied GCPII localization in human and animal tissues under physiological and pathophysiological conditions. However, the data are rather inconsistent, likely for two main reasons. First, different studies used distinct detection methods, ranging from Western blotting, ELISA, and immunohistochemistry to in vivo imaging (17). The data gained from these experiments thus may differ dramatically due to the variable detection limits of each method. Second, various antibodies have been used, and most of them were poorly characterized, which may have led to their improper use and thus generation of false positive/negative results. To provide guidance on how to use anti-GCPII antibodies in the most efficient way for individual immunochemical methods, we recently published a detailed characterization of the most commonly used anti-GCPII antibodies (36).
In contrast to the wealth of data on GCPII, almost no data are available on the distribution of GCPIII within the human body. Notably, several antibodies recognizing GCPII also detect GCPIII (36). Therefore, it is possible that some tissues reported to be GCPII-expressing are in fact also or solely GCPIII-expressing. The major challenge facing determination of GCPIII localization has been the absence of an available antibody selective for native or denatured GCPIII. However, this obstacle was recently overcome by use of a BCG-cleavage assay for specific GCPIII detection in human tissues (24).
Despite inconsistencies in the data, a general consensus has been reached on GCPII expression within the human body: it is expressed in the prostate (5, 37-48), nervous system (39, 45-47, 49), small intestine (39-41, 43-45, 47, 48), and kidney (5, 37, 40, 41, 43, 45-47, 50). Closer examinations of individual tissues using either human or rat histological samples localized GCPII to the acinar epithelium in the prostate (40, 45, 47), astrocytes and Schwann cells in the nervous system (49, 51-53), duodenal mucosa in the small intestine (40, 45), and proximal tubules in the kidney (40, 45, 47, 50). In addition, GCPII has been repeatedly detected in human blood (54-57), and we have observed pronounced GCPII expression in the salivary glands (unpublished observation). Several studies also suggest that GCPII expression is not restricted to the sites mentioned above, but that the enzyme is to a lesser extent ubiquitous throughout the human body, expressed in tissues such as the breast, colon, heart, liver, ovary, testis, urinary bladder, and uterus. (44-47).
Only one report has addressed localization of GCPIII in humans (24). Among the tissues examined (testis, ovary, placenta, spleen, prostate, pancreas, heart, jejunum, kidney, and brain), GCPIII was detected mainly in the testis and to a much lesser extent in the prostate, brain, kidney, and small intestine. Nevertheless, a more rigorous investigation using a broader set of human tissues and more biological replicates is needed.
While GCPIII has not been associated with any pathophysiological condition to date, GCPII has been linked to several different diseases. In particular, GCPII is recognized as a prostate cancer marker because its high expression within prostate tumor cells has been reported repeatedly (5, 37-41, 43-46, 58-61). Moreover, the level of GCPII expression increases with increasing prostate cancer grade (58-60), and a substantial amount of GCPII is present in different prostate cancer metastases (5, 38, 40, 59, 62).
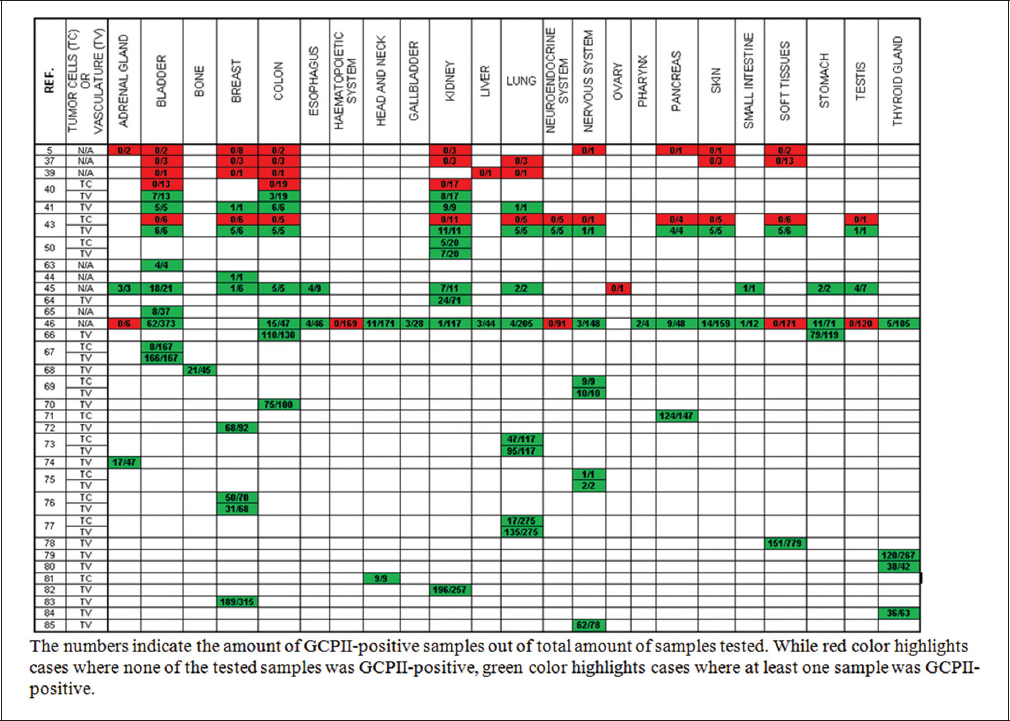
Although the first studies assessing GCPII expression in malignancies suggested that it is expressed only in prostate cancer (5, 37, 39), it has since been detected in numerous other cancers. These include bladder carcinoma, breast carcinoma, colorectal adenocarcinoma, non-small cell lung carcinoma, glioma, clear cell renal cell carcinoma, and thyroid carcinoma (for a complete list, see Table 1) (40, 41, 43-46, 50, 63-85). Nevertheless, while GCPII is localized mainly in prostate cancer tumor cells (38, 40, 58-60), its expression in most other cancers is restricted to the tumor-associated neovasculature (40, 41, 43, 50, 66, 86).
Recently, overexpression of GCPII was reported in inflammatory bowel disease (IBD) (87). FOLH1 gene upregulation in IBD had been demonstrated previously (88-90), and GCPII expression was confirmed in one case using immunohistochemistry (89). In the recent study, researchers explored whether GCPII is present in an active form in the affected intestinal mucosa of IBD patients. They reported a 3- to 40-fold increase in GCPII enzymatic activity compared to colon samples from healthy volunteers (87). The significance of this increase remains to be resolved but the findings suggest that GCPII could serve as a biomarker for IBD.
Although GCPII is expressed in many tissues, its function has been established only in the brain and small intestine. The functions in both tissues are distinct but closely correlate with the known enzymatic activities of GCPII. To study the physiological role of GCPII in more detail, several independent research groups attempted to generate GCPII null mutant mice, with conflicting results. While some reported that GCPII deficiency is embryonically lethal, others demonstrated that inactivation of the Folh1 gene generates viable GCPII null mutant mice with no obvious phenotype (91-96). Investigations of viable GCPII null mutant mice have focused mainly on the nervous system (91, 94, 97, 98). Further examination of other tissues is thus desirable, as it could confirm or disprove the non-enzyme functions of GCPII suggested in the literature, as well as point out yet-to-be-revealed physiological roles of GCPII.
The physiological function of GCPIII has not yet been determined. Because GCPIII possesses similar enzyme activities as GCPII, it has been suggested that GCPIII serves as an enzyme complementary to GCPII. This hypothesis was further strengthened by the fact that NAAG-hydrolyzing activity was detected in the brains of GCPII null mutant mice (91). Nevertheless, considering that the expression level of GCPIII in the testis is more than one order of magnitude higher than that of GCPII, it is highly probable that GCPIII also fulfills other functions in the human body (24).
In the brain, GCPII functions as a neuromodular enzyme by cleaving and thus deactivating the most abundant peptide neurotransmitter, NAAG (99). During synaptic transmission, NAAG is released together with other neurotransmitters from the presynaptic neuron into the synaptic cleft and participates in downregulation of postsynaptic neuron stimulation (99-101). This regulation may be executed through two different types of receptors capable of NAAG binding: N-methyl-D-aspartate receptors (NMDARs) and metabotropic glutamate receptors (mGluRs) (102, 103).
The physiological relevance of the interaction of NAAG with NMDARs has been debated in the literature (104). Moreover, a clear consensus has not been reached on which action within the synaptic cleft this interaction would evoke (105). On the other hand, the pathway of NAAG action through mGluRs has been established and seems to be generally accepted (104, 105) (Figure 6). Once released into the synaptic cleft, NAAG activates type 3 metabotropic glutamate receptors (mGluR3) located at the membranes of both presynaptic neurons and astrocytes (106-108). The subsequent events depend on the site of action. While stimulation of presynaptic mGluR3 inhibits additional release of neurotransmitters from the presynaptic neuron (109-112), activation of mGluR3 on astrocytes triggers a signaling pathway that results in the production and secretion of neuroprotective growth factors (113-115). Both these actions help prevent excessive nerve stimulation that could lead to pathological effects. Because GCPII serves as a NAAG deactivator, it is not surprising that substantial efforts have been made to identify a selective GCPII inhibitor for potential use in the treatment of neurological disorders (for more information see Section 6.2.).
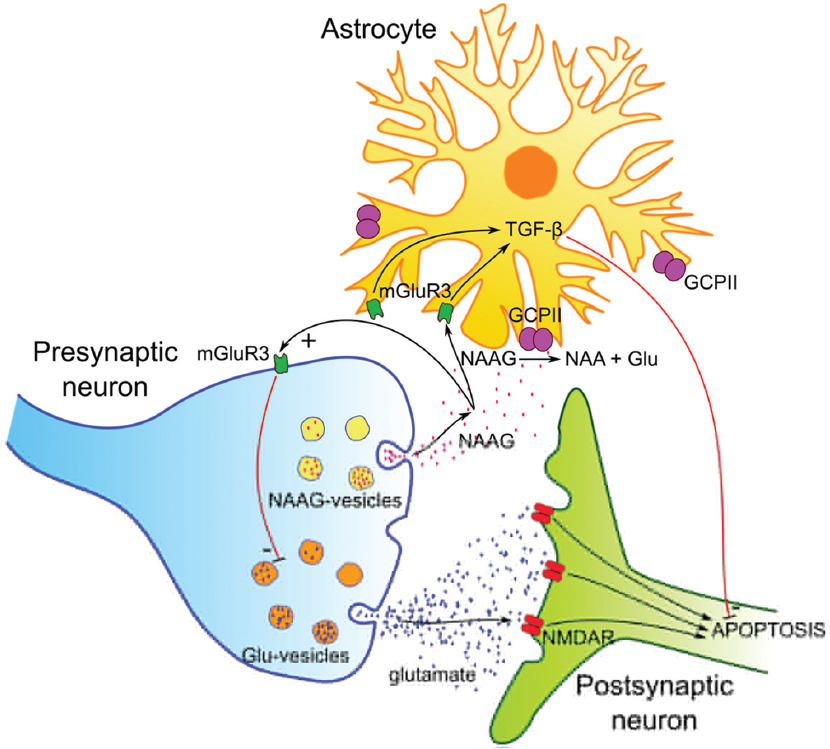 Figure 6
Figure 6Proposed functions of NAAG, glutamate and GCPII on synaptic terminals. The neurotransmitters NAAG and glutamate are released into the synaptic cleft from a presynaptic neuron. Glutamate activates N-methyl-D-aspartate receptors (NMDAR), which leads to apoptosis under pathological conditions associated with excessive glutamate levels. NAAG activates metabotropic glutamate 3 receptors (mGluR3) on presynaptic neurons (resulting in the decrease of released glutamate) and on astrocytes (leading to the secretion of neuroprotective factors, such as TGF-β). GCPII (and possibly GCPIII) hydrolyzes NAAG into N-acetyl-L-aspartyl (NAA) and glutamate and consequently abolishes the protective effects of NAAG.
In the small intestine, GCPII is involved in folate absorption. Natural folates occur in the diet as poly-gamma-glutamates (116-118). The process of natural food folate absorption comprises two basic steps. First, GCPII located in the brush border membrane of the small intestine consecutively cleaves polyglutamylated folates to produce monoglutamylated forms of folate (119). Second, monoglutamylated folates are taken up by intestinal cells through the proton-coupled folate transporter (120). This transporter has been shown to be highly specific for monoglutamylated folates (121). GCPII thus plays a crucial role in folate absorption. For this reason, several research groups have studied the possible influence of FOLH1 gene polymorphisms on folate metabolism, but the data are rather conflicting (122-135).
One of the two naturally occurring polymorphisms reported to date—rs61886492, also known as C1561T—has been associated with increased serum folate levels (123, 126, 127, 130-132). While two reports also showed an association between rs61886492 and decreased serum homocysteine levels (130, 132), others failed to confirm this link (123, 126, 127, 131). Moreover, several studies did not observe any effect of rs61886492 on serum folate or homocysteine (124, 125, 128, 129). At the protein level, the rs61886492 mutation results in the H475Y mutation (122), which does not alter GCPII activity towards polyglutamylated folates (26). If this polymorphism is indeed involved in altered serum folate levels, the mechanism of action does not directly relate to the enzymatic activity and remains to be determined.
The second naturally occurring FOLH1 polymorphism—rs202676, also known as T484C—has been studied to a much lesser extent. Nevertheless, recent reports have shown that this polymorphism correlates with lower red blood cell folate levels (133-135), which is in accordance with the predicted hypofunction of GCPII caused by this missense mutation (133). Indeed, structural modeling suggested that the corresponding protein mutation Y75H leads to alteration in the secondary and tertiary structure of GCPII and thus affects the binding of polyglutamylated folates (133).
Numerous studies have addressed the possible association of FOLH1 gene polymorphisms with the incidence of various pathological conditions resulting from folate metabolism dysregulation. These include severity of negative symptoms in schizophrenia (136), neural tube defects (125, 126, 133, 137-140), risk for different cancers (128, 132, 141-144), and cardiovascular disease risk (123). No association with colorectal (128, 132, 141), gastric (142), and lung (143) cancer risk or cardiovascular disease risk (123) was observed. Interestingly, one study suggested that rs61886492 might have a protective role in breast cancer; however, the cohort consisted of only 68 women from one ethnic group (144). Neural tube defects are the best-studied pathological condition in relation to FOLH1 gene polymorphism. However, studies of this correlation are rather inconsistent since one report showed that FOLH1 gene polymorphism is potential risk factor for neural tube defects (133) while others failed to find this association (125, 126, 137, 139, 140). Further research is thus necessary to shed the light on the possible involvement of FOLH1 gene polymorphisms in neural tube defects.
The first GCPII null mutant mice were described in 2002 by Bacich and co-workers (91). These mice were prepared by deletion of intron-exon boundary sequences of exons 1 and 2 of the Folh1 gene and insertion of several in-frame stop codons between exon 1 and 2. The resulting GCPII null mutant mice expressed neither a GCPII mRNA nor a shortened version of the protein, and NAAG cleavage activity was dramatically decreased in the brain and kidney of these mice compared to wild-type (WT) mice. Interestingly, the levels of NAAG and glutamate in the brain were not significantly altered in the GCPII null mutant mice. The authors thus hypothesized that deletion of the Folh1 gene may induce changes in expression of other genes, including Naalad2 (which encodes GCPIII). Phenotypic examination of the GCPII null mutant mice did not reveal any significant differences in standard neurological behavior in comparison with WT mice. Nevertheless, these GCPII null mutant mice were protected from peripheral neuropathies and traumatic brain injury (TBI) (97).
In 2003, scientists from the research group of Joseph T. Coyle reported that manipulation of embryonic stem cells by deletion of exons 9 and 10 of the Folh1 gene leads to embryonic lethality (92). The same group subsequently tried to reproduce the work of Bacich and co-workers by deleting exons 1 and 2 of the Folh1 gene, but their attempt did not lead to generation of viable GCPII null mutant mice (93).
In 2015, another research group reported preparation of GCPII null mutant mice by deleting exons 3 to 5 of the Folh1 gene (94). The resulting GCPII null mutant mice showed similar phenotypic characteristics as the mice prepared by Bacich and co-workers. While no overt behavioral differences between GCPII null mutant and WT mice were observed, GCPII null mutant mice were less susceptible to TBI and showed improved long-term behavioral outcomes after TBI (94).
Basic information about the generation and phenotyping of GCPII null mutant mice also can be found on the International Mouse Phenotyping Consortium (IMPC) website (95). These viable GCPII null mutant mice were prepared by deleting exon 3 of the Folh1 gene. In contrast to the results of others, GCPII null mutant mice characterized by the IMPC do not show any clear phenotype (95). However, the full set of phenotyping data has not yet been released.
Recently, we also reported generation of viable GCPII null mutant mice using TALEN-mediated disruption of the sequence encoding GCPII active site (96). It remains unclear why neither strategy attempted by Coyle’s research group led to production of live GCPII null mutant mice. However, thanks to the work of four other laboratories, it now seems to be evident that knock-out of the GCPII-encoding gene does not lead to mouse embryonic lethality.
No GCPIII null mutant mice have been reported to date, although generation and investigation of such mice could be very useful. It could not only shed the light on the physiological role of GCPIII but also point out unique physiological roles of GCPII.
Outside of the nervous system and small intestine, GCPII’s role in human physiology remains elusive. Researchers are debating whether GCPII may have other, unrecognized physiological substrates or serve as a receptor for a yet-to-be-identified extracellular ligand. However, not enough data has been published for any hypothesis to be widely accepted by the scientific community. The receptor theory is based on the fact that GCPII possesses sequence and structural similarity to the transferrin receptor (16, 145) and is capable of internalization upon ligand binding (146, 147). One report even suggested that GCPII itself might be able to transport folate into cells (148). However, no follow-up research has been performed to date. The description of GCPII as a folate transporter, which has recently repeatedly appeared in the literature (6, 140, 149), should thus be avoided.
Although GCPII is present in prostate cancer cells and in the neovasculature of many solid tumors, its function in these pathologies is still unclear. Some reports link GCPII to tumor progression and carcinogenesis (148, 150), in accordance with its expression in prostate cancer metastases (5, 38, 40, 59, 62). On the other hand, others suggest that GCPII instead suppresses cancer invasiveness (151). A detailed recent review of GCPII’s potential involvement in malignant processes is available elsewhere (6).
While many studies have aimed to elucidate GCPII function, the physiological role of GCPIII has not been extensively investigated. As GCPIII has been identified as a BCG-hydrolyzing enzyme, it is likely involved in BCG metabolism. However, the physiological role of BCG has been studied by only one research group, and it remains poorly understood. Potentially, BCG might be important for brain development, as it was found in high concentrations in the newborn rat brain but disappeared with maturation (152, 153). Similarly, BCG was detected in the germinal cells of the rat testes, and its amount increased with the appearance of late spermatocytes and early spermatids (154), suggesting it could be involved in spermatogenesis. Follow-up studies linked BCG to physiologically important chelation of iron and copper (155, 156). Indeed, BCG, as a low molecular weight chelator, may play a role in reactive oxygen species-scavenging activities, inhibition of xanthine oxidase, or activation of aconitase (155-157).
Given its overexpression in pathology and its physiological functions, GCPII is considered a promising target for diagnosis and therapy of prostate and other cancers, as well as a target for treatment of neurological disorders and potentially IBD. For potential treatment of neurological disorders and IBD, researchers aim to block GCPII hydrolase activity using selective inhibitors, while in experimental cancer treatments, GCPII serves as a molecular address for targeted delivery of therapeutics. The therapeutic and diagnostic potential of GCPII was recently comprehensively reviewed by Evans and coauthors (6) and Wustemann and coauthors (158). We briefly summarize the current state of the field.
Since inhibition of NAAG-hydrolyzing activity was shown to be neuroprotective in mice, considerable effort has been made to develop potent GCPII/GCPIII inhibitors (159-164). Both enzymes contain two active-site zinc ions, and their S1’ pockets exhibit a strong preference for glutamate/glutamate-like moieties (22). This active site “architecture” led to the development of a canonical GCPII inhibitor structure consisting of a glutamate moiety (fitting into the S1’ subsite) and a zinc-binding group (“chelating” the zinc ions). Based on the nature of the zinc-binding group, the inhibitors are usually categorized into three groups: phosphorus-containing compounds (e.g. phosphonates and phosphinates), ureas, and thiol-based compounds (165, 166). Unfortunately, the majority of GCPII/GCPIII inhibitors identified to date are rather polar compounds with poor bioavailability (17, 167). There is thus a need to identify novel inhibitor scaffolds that are less polar, yet still potent and selective. Recently developed high-throughput assays could be applied for this purpose (168, 169).
Due to the similarity between the GCPII and GCPIII active sites, most inhibitors developed to date bind to both enzymes. While this feature may be beneficial for potential drugs targeting neurological disorders, it could be a pitfall for small molecule-based targeting in cancers, which needs to be highly specific. Furthermore, as the physiological role of GCPIII is unknown, inhibition of this close GCPII homolog could cause unexpected side effects. In particular, the testis, as a high GCPIII-expressing organ, may represent a possible off-target tissue for GCPII-based tumor-targeted drug delivery.
Considering the high similarity of the GCPII and GCPIII active sites, it is not surprising that development of GCPII selective inhibitors is challenging. Nevertheless, Tykvart et al. recently prepared a GCPII inhibitor with a rigid linker that exhibits more than 4,000-fold selectivity for GCPII (Ki = 49 pM) over GCPIII (Ki = 210 nM) (170). The authors also investigated some common GCPII inhibitors, such as DKFZ-PSMA-11 and DCIBzL, and found out that these inhibitors also possess a three-order-of-magnitude selectivity for GCPII over GCPIII (170). However, their Ki values towards GCPIII are in the low nanomolar range (Ki = 13 nM and 3 nM, respectively), due to which these inhibitors may be still capable of interfering with potential GCPIII functions, including BCG metabolism. Overall, the work of Tykvart et al. indicated that selectivity for GCPII over GCPIII can be achieved by developing compounds that bind to GCPII exosites that are not present in GCPIII. In addition, other structural features that differ between GCPII and GCPIII, such as the “entrance lid”, Pro-rich region, and zinc cation occupancies (14), should be considered for the future development of highly selective GCPII inhibitors.
A number of in vivo studies have shown that inhibition of GCPII ameliorates conditions caused by pathologically increased glutamate levels and have demonstrated the potential of selective GCPII inhibitors for treatment of various neurological disorders (reviewed in (165, 171)). These include ischemic and traumatic brain injury (172-175), amyotrophic lateral sclerosis (176), inflammatory and neuropathic pain (164, 177, 178), schizophrenia (179, 180), multiple sclerosis (181), and autoimmune encephalomyelitis (182). The majority of these experiments were carried out in rats and mice, which serve as suitable animal models due to the similarities between rat and mouse GCPII and their human orthologue in the context of neurological diseases (47, 183).
Importantly, GCPII inhibitors do not seem to influence normal glutamate functions. Rather, their effect is confined to suppressing excessive pathological glutamate signaling (172). This offers a potential advantage over other strategies modulating glutamatergic neurotransmission, such as use of NMDAR antagonists, which often cause undesirable side effects (184).
Although several GCPII inhibitors have been successful in animal models (e.g. 2-PMPA, ZJ-43 and 2-MPPA), none has become a clinically used therapeutic agent, in part due to generally poor oral bioavailability and low blood-brain barrier penetration. The thiol-based inhibitor 2-MPPA was evaluated in phase I clinical trials (185) that found it safe and well-tolerated; however, its further development was stopped, likely due to concerns about the adverse properties of thiols (e.g. their nucleophilicity, propensity to oxidation, metabolic instability) (186). Recently, a prodrug strategy has emerged to increase the lipophilicity of GCPII inhibitors and thus improve oral absorption. In this approach, the polar groups necessary for inhibitor binding to the GCPII active site (e.g. phosphonate, carboxylate, and hydroxamate) are masked by hydrophobic moieties (isopropyloxycarbonyloxymethyl, thiolactones) that are cleaved off in the target tissues by hydrolases (167, 187-189).
GCPII enzyme activity has been shown to be elevated in IBD patients (87). In a follow-up study, the researchers reported that systemic administration of the GCPII inhibitor 2-PMPA markedly decreases IBD severity and ameliorates symptoms in mice (87, 190). Even though the mechanism of action has not been determined, these results suggest that GCPII inhibition might contribute to IBD therapy.
The current major focus of GCPII research is imaging and therapy of prostate cancer and its metastases. In addition, given its expression in the neovasculature of many solid tumors, GCPII is being investigated for its potential to serve as an imaging and therapeutic target in other cancers (191). Various techniques are available for molecular imaging of GCPII within malignant tissue. The most frequently used methods are radioligand-based positron emission tomography (PET) and single photon emission computed tomography (SPECT), usually combined with computed tomography (CT) (192, 193).
The only agent approved by the U.S. Food and Drug Administration (FDA) for imaging of prostate cancer is the 111In-labeled monoclonal antibody 7E11 (194, 195), known as 111In-capromab pendetide (trade name ProstaScint™). Second-generation radiolabeled monoclonal antibodies recognizing the extracellular portion of GCPII (such as J591, which enables visualization of viable cells) have been tested in clinical trials (196, 197). The main disadvantage of the antibodies for imaging is half-life being as long as several days, which may cause harm in off-target tissues. From this reason, low-molecular-weight inhibitors have been developed since they are capable of rapid removal from circulation by renal filtration. Urea-based GCPII inhibitors radiolabeled with 18F, 68Ga, and 64Cu are considered the most promising PET tracers for GCPII imaging. These include 68Ga-PSMA-11 (or 68Ga-PSMA-11-HBED-CC; Figure 7A) and 18F-DCFPyL (Figure 7B), which demonstrated potential for identification of metastases and recurrent prostate cancer (198-205). Further research and development of improved and novel GCPII imaging probes is ongoing (206-212).
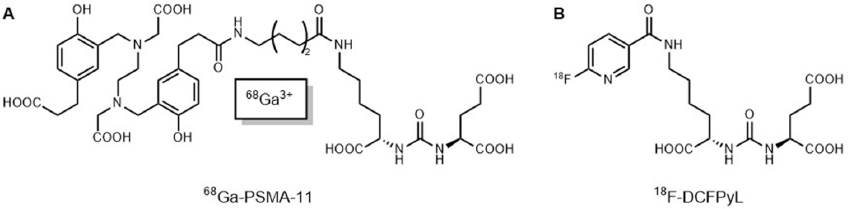 Figure 7
Figure 7GCPII-targeted PET imaging agents. A: Radiometal-labeled agent 68Ga-PSMA-11 (198). B: Radiolabeled agent (18F)DCFPyL (199).
Radioimmunotherapy/radioligand therapy is the most common approach for prostate cancer therapy using GCPII as the target. The first such “therapeutic” agent targeting GCPII was 90Y-7E11 antibody; however, it exhibited hematologic toxicity in clinical trials (213, 214). This toxicity, as well as the inability of 7E11 to bind viable tumor cells, spurred the development of second-generation radiolabeled antibodies such as J591 (215). 177Lu-J591 demonstrated excellent GCPII-targeting in men with metastatic castration-resistant prostate cancer (mCRPC), resulting in a dose-dependent decline in their PSA levels (197, 216, 217). Recently, the “third-generation” of highly potent monoclonal antibodies recognizing GCPII has been introduced, and these represent prime candidates for the development of novel theranostics to target GCPII (218).
Small molecules labeled with therapeutic radionuclides, such as the recently developed 177Lu-PSMA-617, are also promising therapeutic agents for treatment of mCRPC (219). 177Lu-PSMA-617 has been studied intensively during the last two years (220-226). Two recent German retrospective multicenter studies evaluating 177Lu-PSMA-617 radioligand therapy (145 and 59 mCRPC patients) showed that the radioconjugate exhibits favorable safety and efficacy (a ≥ 50% PSA decline occurred in 45% and 53% of patients, respectively) (222, 223). Similar results, showing high response rates, low toxicity and reduction of pain in men with mCRPC, were obtained by a single-arm, single-center, phase 2 trial performed in Australia (224). In February 2017, the first U.S. multicenter phase II trial of 177Lu-PSMA-617 radioligand therapy received FDA clearance (estimated study completion date: April 11, 2019) (225). Quite recently (May 2018), a multicenter randomized phase III study has been launched (estimated study completion date: May 2021), which will compare overall survival in patients with GCPII-positive mCRPC who receive 177Lu-PSMA-617 in addition to best standard of care versus patients treated with best standard of care alone (226).
In addition to radiotherapy, antibodies and small molecules could be used for delivery of cytotoxic molecules and toxins. Deimmunized J591 was conjugated to an extremely potent microtubule-depolymerizing drug, DM-1 (227); unfortunately, the conjugate exhibited neurotoxicity and limited activity in mCRPC patients (likely due to the deconjugation of DM-1) (228). More encouragingly, a conjugate of an anti-GCPII antibody and the potent antimitotic drug monomethyl auristatin E exhibited low toxicity and a ≥ 50% PSA decline in 33% of mCRPC patients (229-231). Other conjugates, including a conjugate of J591 and ricin A-chain and a conjugate of the anti-GCPII single-chain antibody fragment D7 and the toxic domain of Pseudomonas exotoxin A (PE40) (232, 233), have not yet been tested in a clinical setting. Nevertheless, the latter significantly inhibited growth of a GCPII-positive tumor xenograft in mice (234). In addition, many small selective GCPII inhibitors conjugated with cytotoxic agents have been prepared (158, 235), with one already being investigated in a phase I clinical trial for the treatment of mCRPC (236).
Other GCPII-targeted drug delivery systems include anti-GCPII aptamers (237-243), polymer-based nanocarriers (244-249), liposomes with co-entrapped drugs (250-252), and nanoparticles targeting GCPII, often designed as theranostics (253-255). We have recently developed structurally diverse nanoparticles (polymer conjugates, nanodiamonds, and virus-like particles) decorated with GCPII inhibitors, which could be used for future drug delivery (256, 257).
Finally, there is an increasing number of reports describing GCPII-based immunotherapy of prostate cancer, although only a few studies have been performed in a clinical setting. These investigational therapies are based on dendritic cells (258, 259), GCPII-CD3 diabodies and Fab conjugates (260, 261), cytotoxic T lymphocytes (262), T-cells bearing a chimeric antigen receptor (263-266), PEI-PEG-DUPA conjugates targeting polyinosine/polycytosine acid inducing tumor regression (267), and DNA-encoded anti-GCPII monoclonal antibodies mediating antibody-dependent cellular cytotoxicity (268).
Despite decades of research, the scientific community remains intensely interested in GCPII. A Pubmed query using GCPII/PSMA as a search term returns almost 800 entries from 2017 to present, most dealing with GCPII as a molecular address for prostate cancer theranostics. This approach leverages molecular recognition of GCPII by monoclonal antibodies, nanobodies, nucleic acid aptamers, or small-molecule inhibitors for the specific delivery of nanoparticles (polymer conjugates, nanodiamonds, virus-like particles) decorated by cytotoxic drugs, radiolabels, or fluorescent probes for efficient cancer treatment and imaging. Dozens of papers describing novel antibody conjugates or nanoparticle formulations are published almost every week. Regardless, very little is known about the role of GCPII in cancer development, both in the prostate and in the neovasculature of other solid tumors. The proteolytic function of GCPII is almost certainly not crucial for its putative role in cancer development, as inhibition of GCPII alone has no anticancer activity. Potentially, GCPII could serve as a receptor of a yet-to-be-identified signaling ligand contributing to cell growth or migration. Additionally, the physiological function of GCPII outside the brain and jejunum remains unknown, as does the possible role of GCPII in development and pathogenesis of IBD. Furthermore, while GCPII is highly expressed in the kidneys, there is no hypothesis for its role in this organ.
Even less is known about the function of GCPIII. It has been long viewed as a mere isoenzyme of GCPII, not distinguishable by its activity or by immunochemical methods. Only recently it was established that this enzyme is highly expressed in the testis, where it co-localizes with its most specific cognate substrate, BCG. These findings also raise the need for further research. The role of BCG and its degradation in the testis is yet to be determined, as is the molecular mechanism by which GCPIII specifically recognizes BCG while GCPII does so much less effectively. A structural analysis of the complex of GCPIII with BCG completed by QM/MM/MD calculations could help explain the specificity of the enzyme and the role of metals in this particular enzymatic activity. It is difficult to imagine that such a high concentration of an enzyme and specific cognate substrate in the same tissue does not have an important physiological consequence. To elucidate the physiological function of the BCG-hydrolyzing activity of GCPIII, specific novel tools will be required, such as GCPIII-specific antibodies or selective inhibitors of this homolog. The distinct structural features of GCPIII discussed in this review could be considered to achieve this task.
Null mutant animals likely represent the most direct avenue to addressing these unanswered questions. Some attempts to create GCPII null mutant mice resulted in embryonic lethality, while others led to viable mice with no clear phenotype. Our own animal experiments support the latter finding – GCPII null mutant mice develop normally with minor phenotypic characteristics appearing in the later adult stage. GCPIII is most likely capable of complementing the lost GCPII activity in these experimental animals. Development of GCPIII null mutant mice and GCPII/III double null mutant mice would be key to elucidating the function of both homologs. Improving our understanding of the physiological function of these fascinating enzymes will hopefully lead to their better use as tools for future diagnostic and therapeutic approaches.
The authors would like to acknowledge Dr. Hillary Hoffman for language editing. This work was supported by InterBioMed project LO 1302 from the Ministry of Education of the Czech Republic.
Abbreviations: ABS: arene-binding site, BCG: β-citryl-L-glutamate, FDA: Food and Drug Administration, FolGlun: polyglutamylated folates, GCPII: glutamate carboxypeptidase II, GCPIII: glutamate carboxypeptidase III, IBD: inflammatory bowel disease, IMPC: International Mouse Phenotyping Consortium, mGluRs: metabotropic glutamate receptors, mCRPC: metastatic castration-resistant prostate cancer, NAA: N-acetyl-L-aspartyl, NAAG: N-acetyl-L-aspartyl-L-glutamate, NAAG2: N-acetyl-L-aspartyl-L-glutamyl-L-glutamate, NAALADase: N-acetylated alpha-linked acidic dipeptidase, NMDARs: N-methyl-D-aspartate receptors, PET: positron emission tomography, PSMA: prostate-specific membrane antigen, TBI: traumatic brain injury, WT: wild-type