
- Academic Editor
Hypertrophic cardiomyopathy (HCM) is a prevalent cardiac disease characterized by marked phenotypic variability. Recent advances in diagnosis and treatment have allowed a personalized approach to the treatment of this disease. Depending on the predominant phenotype, management can be tailored to address left ventricular outflow tract obstruction, heart failure, arrhythmia control, and/or sudden cardiac death prevention. This review highlights recent advances that have transformed the therapeutic landscape of HCM. Modern imaging techniques have improved sudden cardiac death risk stratification. The development of myosin inhibitors represents a paradigm shift in the treatment of symptomatic obstructive HCM. Invasive septal reduction techniques have also evolved, with novel approaches such as percutaneous intramyocardial septal radiofrequency ablation and transapical beating-heart septal myectomy. Finally, gene-targeted therapies including replacement, editing and silencing approaches, are emerging as promising strategies for HCM management.
Hypertrophic cardiomyopathy (HCM) constitutes the most frequently inherited
cardiomyopathy with an estimated adult prevalence of 0.2% [1]. HCM in adults is
defined by a left ventricular (LV) wall thickness
Since the first descriptions of obstructive HCM (oHCM) and non-obstructive HCM (nHCM), there has been a revolution in the understanding of its pathophysiology [4, 5]. This has led to substantial changes in diagnosis, treatment, and SCD risk assessment [1]. Advances in imaging techniques, especially cardiac magnetic resonance (CMR), have provided key insights [6]. New risk assessment tools and algorithms have been developed [7]. Myosin inhibitors (MI) have transformed the treatment algorithm of patients with oHCM; being the first pharmacological group to act upon HCM’s pathophysiological mechanism [8]. Many interventional alternatives to Morrow classical septostomy have emerged [9]. Finally, better understanding HCM’s underlying genetic mechanisms has allowed the development of gene therapy (GT), raising the question of whether some HCM patients may be definitively cured in a near future [10].
All of the previous advances, has increased the need for clinicians to be fully updated to offer the best personalized integral management to HCM patients.
Annual SCD in HCM is estimated to be around 0.5–0.8% in adults and 1.2–1.5% in children. It may occur as the initial presentation of HCM [2, 11], being ventricular tachycardia/fibrillation (VT/VF) the most common causes of SCD. Implantable cardioverter-defibrillators (ICD) are an effective therapy for managing life-threatening ventricular arrhythmias [12]. Nonetheless, risk stratification is still challenging and controversial [13].
Both ESC European Society of Cardiology guidelines (ESC) and American Heart Association guidelines (AHA) guidelines [1, 14] uniformly recommend, with class I indication,
ICD implantation in secondary prevention. However, they diverge in their
recommendations for primary prevention (Fig. 1). ESC guidelines endorse the use
of the HCM Risk-SCD model for adults and the HCM Risk-Kids tool for patients
under 16 to estimate 5-year SCD risk [7, 15]. ICD implantation is based on
defined risk thresholds (low risk
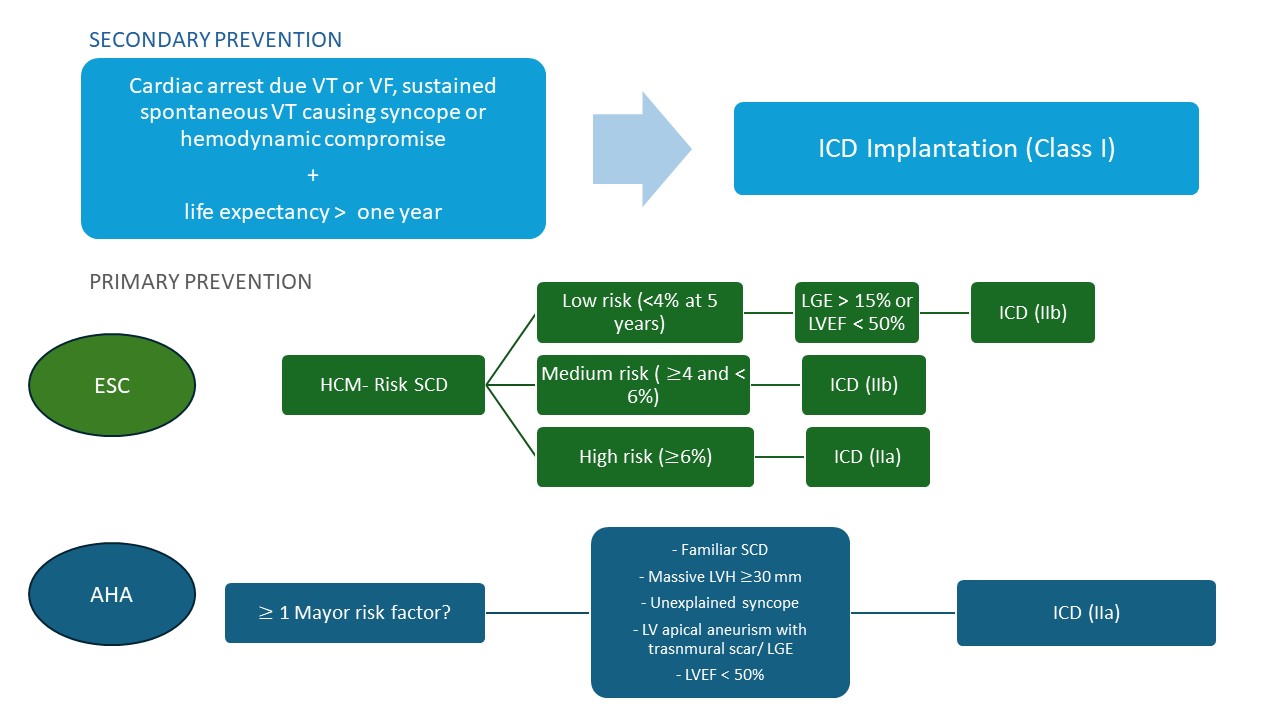 Fig. 1.
Fig. 1.
Flowchart of sudden cardiac death risk assessment and implantable cardioverter-defibrillator implantation indications according to European (2023) and American (2024) guidelines. AHA, American Heart Association guidelines; ESC, European Society of Cardiology guidelines; HCM, Hypertrophic Cardiomyopathy; ICD, Implantable Cardioverter-Defibrillator; LGE, Late Gadolinium Enhancement; LV, Left Ventricular; LVEF, Left Ventricular Ejection Fraction; LVH, Left Ventricular Hypertrophy; SCD, Sudden Cardiac Death; VF, Ventricular Fibrillation; VT, Ventricular Tachycardia.
Key differences exist between guidelines regarding the role of CMR, apical
aneurysm [16, 17], and left ventricular ejection fraction (LVEF) [18, 19] (Table 1, Ref. [1, 14]). While the ESC guidelines view the previous as risk modifiers,
emphasizing the limitations of current evidence and the need for individualized
clinical interpretation, AHA guidelines consider them as independent risk factors
that warrant ICD implantation. These differing strategies result in varying ICD
implantation rates. The AHA method offers a higher sensitivity (
| Risk factor | ESC Guidelines (2023) [1] | AHA Guidelines (2024) [14] |
| Family history of SCD | SCD in |
SCD in |
| Not independently associated with prognosis in pediatric HCM | ||
| Unexplained syncope | Recent ( |
Events |
| Age | Younger patients ( |
Not explicitly discussed |
| Age affects marker sensitivity | ||
| Maximum LV wall thickness | Highest risk if |
|
| Pediatric: z-score | ||
| Left atrial diameter | Larger size linked to increased SCD risk | Not mentioned |
| LV apical aneurysm | Not listed | Regardless of aneurysm size |
| LVEF | Not emphasized | LVEF |
| LVOTO | Uncertain impact of provocable LVOTO or treatment | Not mentioned |
| Conflicting pediatric data | ||
| NSVT | Independent risk factor | Higher risk if frequent ( |
| Exercise-related NSVT may increase risk | ||
| Frequency/duration not clearly predictive | ||
| LGE | Extensive LGE |
HCM, Hypertrophic Cardiomyopathy; LGE, Late gadolinium enhancement; LV, Left Ventricle/Left Ventricular; LVEF, Left ventricular ejection fraction; LVOTO, Left Ventricular Outflow Tract Obstruction; NSVT, Non-Sustained Ventricular Tachycardia; SCD, Sudden Cardiac Death.
Previous ESC and AHA SCD risk stratification models rely on regression analysis methods. However, emerging data derived from CMR, as well as genetic information, are becoming increasingly relevant for identifying arrhythmogenic risk. Looking ahead, new risk stratification paradigms that integrate old and new risk markers are needed. Machine learning and artificial intelligence (AI) offer the potential to synthesize complex data, delivering a more personalized risk assessment, particularly in patients with intermediate or uncertain risk profiles under conventional models.
CMR enhances SCD risk prediction in HCM by providing detailed myocardial tissue
characterization. Parametric mapping techniques like late gadolinium enhancement
(LGE), T1/T2 mapping, and extracellular volume (ECV) to quantify fibrosis, edema,
and extracellular expansion linked to arrhythmic risk can be used [6]. An LGE
affecting
Taking into account these findings, both ESC/AHA guidelines support the use of CMR for SCD risk stratification in low to intermediate risk HCM patients [1, 14]. However, several uncertainties temper the integration of CMR clinical practice [24]. Quantification of LGE varies widely between centres, scans and software programmes, lacking standardization. Moreover, availability of CMR in low resource settings is limited. Before generalization of the recommendations, a consensus on how to incorporate these variables into decision algorithms is required.
Genetic testing may provide prognostic insight into HCM. Carriers of pathogenic or likely pathogenic sarcomere gene variants generally face higher risks of adverse outcomes. Certain mutations and regions in genes such as MYBPC3, TNNT2 and MYH7 have been linked to a worse prognosis and increased arrhythmic risk [25, 26, 27].
Despite the previous guidelines, the most recent 2023 ESC guidelines have removed the presence of a single sarcomeric pathogenic variant as a standalone indication for ICD implantation in patients with intermediate SCD risk. Genetic findings are supportive but not determinative of eligibility [1]. This is due to association inconsistency across all patient groups. Moreover, additional factors such as polygenetic variants and diastolic blood pressure influence disease expression [28], making it challenging to attribute SCD risk to a single variant or genomic region alone [29]. Before integration into clinical algorithms, more investigation is needed in order to correctly delineate genotype–phenotype correlations in HCM.
In the years ahead, AI is poised to become a new tool for SCD risk assessment. It has proven useful in HCM screening through ECG analysis, with high specificity and negative predictive value [30, 31]. Although a validated AI tool specifically designed for SCD risk assessment in HCM is not yet available, several promising models exist. These include AI algorithms that identify high-risk ECG patterns, integrating clinical, genetic and imaging data to predict outcomes [32, 33, 34]. Model generalizability is however hindered in HCM due to limited sample sizes, variable disease expression, and complex phenotypes.
MI are a new drug group, which selectively and reversibly inhibit cardiac myosin adenosine triphosphatase. They stabilize the super-relaxed state of myosin, thus reducing actin-myosin bridge formation and, consequently, the excessive myocardial contractility associated with HCM pathophysiology. This leads to a reduction in LVOTO and LV filling pressures. It also reduces myocardial energy demands and diastolic dysfunction [8, 35] (Fig. 2).
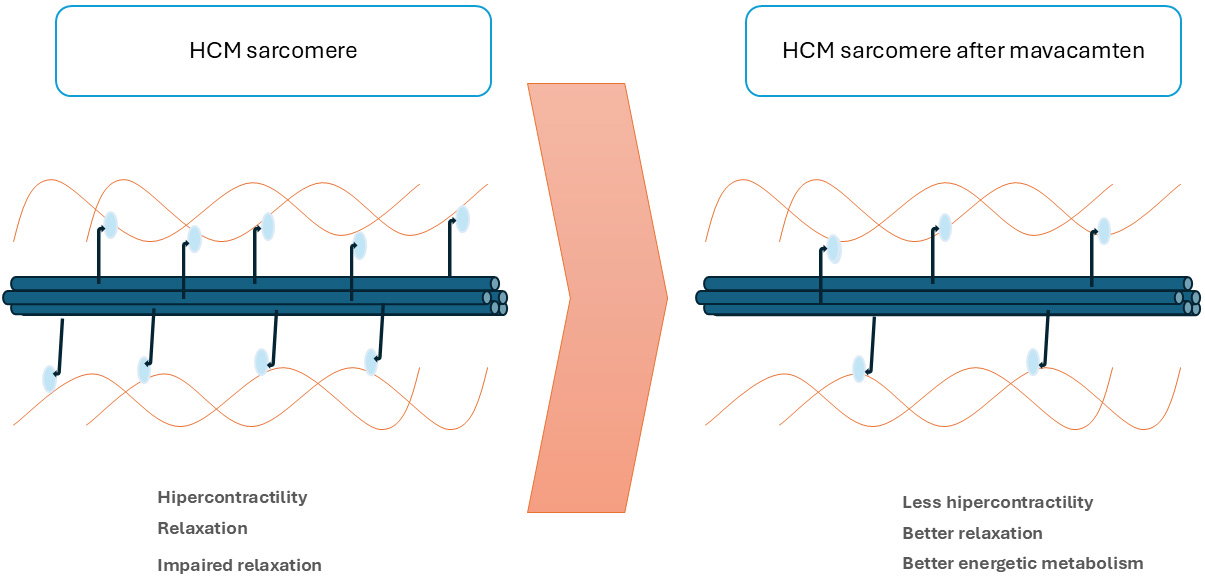 Fig. 2.
Fig. 2.
Mechanism of action of myosin inhibitors. HCM, Hypertrophic Cardiomyopathy.
To date, there are two MI: mavacamten and aficamten. The main differences between them are shown in Table 2. Aficamten, a second-in-class allosteric MI was developed to improve the pharmacokinetic and pharmacodynamic properties of mavacamten. Their most frequent adverse reactions are dizziness (17%), dyspnea (12%), left ventricular systolic dysfunction (5%) and syncope (5%) [1, 35].
| Mavacamten | Aficamten | |
| Half-life (days) | 6–23 | 2.8 |
| Drug interactions | CYP450 | None |
| Dosage titration | Slow (4 weeks) | Quick (2 weeks) |
| Risk of LVEF |
+++ | + |
| Efficacy in oHCM |
LVEF, Left Ventricular Ejection Fraction; oHCM, Obstructive Hypertrophic Cardiomyopathy. +++: higher risk; +: risk but less than with mavacamten.
Mavacamten and aficamten have demonstrated efficacy in oHCM across several trials (Table 3, Ref. [36, 37, 38, 39, 40, 41]). Mavacamten received FDA approval in 2022, while aficamten received approval in 2024. When published, the ESC guidelines were unable to recommend the use of MI as first-line medical therapy, due to the absence of direct head-to-head comparisons with other treatments available at the time. However, they did consider the evidence sufficiently robust to support their use as second-line therapy when optimal medical therapy with beta-blockers, calcium antagonists, and/or disopyramide was ineffective, poorly tolerated or contraindicated [1]. MI may be co-administered with beta-blockers or calcium antagonists. However, safety with negative ionotropic drugs (such as disopyramide) has not been established.
| Study title | EXPLORER-HCM [36] | VALOR-HCM [37] | MAVA-LTE [38] | SEQUOIA-HCM [41] | FOREST-HCM [39] | MAPLE-HCM [40] |
| Drug | Mavacamten | Mavacamten | Mavacamten | Aficamten | Aficamten | Aficamten |
| Design | Double-blind Randomized | Double-blind | Open-label | Double-blind | Open-label | Double-blind |
| Randomized | Extension | Randomized | Extension | Randomized vs metoprolol | ||
| Placebo controlled, Cross-over at week 16 | Placebo controlled | |||||
| N | 251 | 112 | 231 | 282 | 213 | 175 |
| Duration (weeks) | 30 | 56 | 260 | 24 | Ongoing. 48 week analysis. | 24. Final results pending publication. |
| NYHA class | II–III | III–IV | II–III | II–III | II–III | II–III |
| Primary endpoint | pVO2 increase |
Proportion of patients undergoing SRT or remaining guideline elegible ( |
Safety (☺) | pVO2 | Safety (☺) | pVO2 ( |
| Secondary endpoints | LVOT gradient ( |
LVOT gradient ( |
LVOT gradient ( |
LVOT gradient ( |
LVOT gradient ( |
LVOT gradient |
| KCCQ-CSS ( |
NYHA ( |
NT-proBNP ( |
NYHA ( |
NYHA ( |
NYHA | |
| NT-proBNP ( |
NYHA ( |
KCCQ-CCS ( |
NT-proBNP ( |
NT-proBNP | ||
| hs-cTnI ( |
hs-cTnI ( |
KCCQ-CCS | ||||
| KCCQ-CCS ( |
LV mass, LAVI |
CPET, Cardiopulmonary Exercise Test; CV, Cardiovascular; cTnI, High-Sensitivity
Cardiac Troponin I; EF, Ejection Fraction; KCCQ-CSS, Kansas City Cardiomyopathy
Questionnaire–Clinical Summary Score; LVOT, Left Ventricular Outflow Tract;
LVOT-G, Left Ventricular Outflow Tract Gradient; N, Patient Number; NRS,
Numerical Rating Scale; NYHA, New York Heart Association; NT-proBNP, N-Terminal
Pro-B-Type Natriuretic Peptide; pVO2, Maximum Oxygen Consumption; SRT,
Septal Reduction Therapy; HCM, Hypertrophic Cardiomyopathy. ☺: Positive Clinical Trial Results.
In the EXPLORER-HCM trial, mavacamten improved functional capacity, symptoms, and quality of life. 37% of patients on treatment reached the primary endpoint, compared to 17% on placebo [36]. In VALOR-HCM, mavacamten showed a 77% reduction in the need for septal reduction therapy (SRT). In addition, improvements in functional class, LVOT gradients, and quality of life were observed [37]. Similarly, the SEQUOIA-HCM trial of aficamten reported increased peak VO2, significant reductions in LVOT gradients, better New York Heart Association (NYHA) class and improvement in symptom scores. The time that patients remained eligible for SRT shortened by 78 days [37].
Regarding long-term data, MAVA-LTE [38] is the five-year long-term study of
mavacamten and FOREST-HCM [39] was the extension trial of aficamten. Both trials
assessed long-term safety and tolerability, and included individuals who enrolled
in previous pivotal studies. No treatment-related serious adverse events were
identified. In MAVA-LTE, mean LVEF decreased by 11% from baseline to week 180,
but remained within the normal range. In both studies, sustained reductions in
LVOT gradient, and improvements in functional and hemodynamic parameters were
observed during follow up. More real-world data from the Risk Evaluation and
Mitigation Strategy (REMS) program support the safety and effectiveness of
mavacamten. In 70% of patients, LVOT gradient reductions to
Although not currently approved for use in children, pediatric studies with MI are underway. SCOUT-HCM [43] and CEDAR-HCM [44] are two currently recruiting clinical trials designed to evaluate the efficacy, safety, and pharmacokinetics of mavacamten in adolescents with symptomatic oHCM. Endpoints include, amongst others, changes in LVOT gradient, NYHA functional class and cardiac biomarkers. CEDAR-HCM will recruit children aged 6 to 11 years in its open-label extension study.
MAPLE-HCM is the first head-to-head trial comparing MI with beta-blockers. Preliminary results demonstrate that aficamten significantly improved peak VO2, with a favorable safety profile [40]. It remains to be determined if evidence will be sufficient to propose MI use as first-line monotherapy in oHCM.
A shared underlying abnormality, regardless of hemodynamic features, in both oHCM and nHCM, has led to the clinical trials designed to assess MI efficacy in patients with nHCM (Table 4, Ref. [45, 46, 47]).
| MAVERICK-HCM [45] | ODYSSEY-HCM [46] | ACACIA-HCM [47] | |
| Drug | Mavacamten | Mavacamten | Aficamten |
| Design | Double-blind | Double-blind | Double-blind |
| Randomized | Randomized | Randomized | |
| Placebo-controlled | Placebo controlled | Placebo-controlled | |
| N | 59 | 580 | 420 |
| Duration (weeks) | 16 | 48. Final results pending publication. | Ongoing. 72. Results expected in 2026 |
| NYHA class | II–III | II–III | II–III |
| Primary endpoint | Safety (☺) | KCCQ-CSS (-) | KCCQ-CSS |
| pVO2 (-) | |||
| Secondary endpoints | pVO2 + NYHA (=) | VE/VCO2 | pVO2, Ve/VCO2 |
| NT-proBNP and hs-cTnI ( |
NYHA | NYHA | |
| NT-proBNP | LAVI | ||
| KCCQ-CSS | NT-proBNP | ||
| MACE |
Hs-cTnI, High-Sensitivity Cardiac Troponin I; KCCQ-CSS, Kansas City
Cardiomyopathy Questionnaire–Clinical Summary Score; LAVI, Left Atrial Volume
Index; MACE, Major Adverse Cardiovascular Event; pVO2, Maximum Oxygen
Consumption; VE/VCO2, Ventilatory Equivalent for Carbon Dioxide. ☺: Positive Clinical Trial Results; -: Negative Clinical Trial Results; =: indicates no significant change.
Phase 2, MAVERICK-HCM [45] included 59 patients with symptomatic nHCM. Its
primary outcome was safety. Mavacantem was well tolerated, with no differences in
reported serious adverse events. Drug discontinuation because of LVEF reduction
to
However, phase 3 ODYSSEY-HCM refuted MI clinical utility in nHCM [46]. Including
580 patients with nHCM in NYHA functional class II–III, it did not meet its
primary outcome. No significant improvement was observed in Kansas City
Cardiomyopathy Questionnaire–Clinical Summary Score (KCCQ-CSS) or in pVO2 after
48 weeks of treatment. Full results are still to be published. ACACIA-HCM [47] is an ongoing nHCM clinical trial assessing aficamten. It will include 420
patients and its primary endpoint is a change in KCCQ-CSS from baseline to week
36. Secondary endpoints are changes in maximal pVO2 and submaximal (Ve/VCO2)
exercise capacity, proportion of patients with
Small sub-studies using both echocardiography and CMR suggest that MI can promote favorable myocardial remodeling. Treatment has been associated with regression of septal wall thickness and indexed LV mass, reduction in left atrial volume, and improvement in diastolic filling patterns. Modest regression of fibrosis has also been observed through LGE and T1 mapping. Some patients showed normalization of previously abnormal ECGs [41, 48].
In the EXPLORERHCM CMR sub-study, after 30 weeks of mavacamten, the mean left ventricular mass index (LVMI) was decreased by 17.4 g/m2. The LAVI was also decreased by 10.3 mL/m2. There were no significant differences in LGE or ECV between groups at follow-up [48]. Similarly, in the SEQUOIA-HCM CMR sub-study, patients experienced reductions in LVMI (–15 g/m2) and LAVI (–13 mL/m2). Maximal wall thickness (–2.1 mm) and indexed extracellular volume mass (–3.9 g/m2) also decreased. Replacement fibrosis, assessed by LGE, remained stable with no statistically significant differences [41]. These findings may indicate that MI do more than just relieve symptoms. They may reverse key structural features of HCM phenotype, especially if treatment is initiated at earlier stages.
Negative results on the use of MI in nHCM may reflect fundamental pathophysiological differences between both HCM phenotypes. While obstruction drives symptoms in oHCM, diastolic dysfunction, caused by impaired myocardial bioenergetics, appears to be the main issue in nHCM. New treatment strategies specifically targeting nHCM are therefore needed.
IMPROVE-HCM evaluated ninerafaxstat, a cardiac myotrope in 67 patients with nHCM. The trial’s primary endpoint was safety and tolerability over 6 weeks. Secondary efficacy outcomes included ventilatory efficiency (VE/VCO2) and quality of life (KCCQ-CSS). Although results were promising, a phase 3 trial is yet to be initiated [49].
Potential use of sodium-glucose co-transporter 2 inhibitors (SGLT2i) in nHCM has
also been evaluated. A clinical trial tested SGLT2i in patients with diabetes and
symptomatic nHCM with preserved ejection fraction (EF). Its primary composite
endpoint (a
The 2023 ESC guidelines recommend SRT for patients with oHCM who have severe
symptoms and a LVOT gradient
For decades, septal myectomy and alcohol septal ablation (ASA) have represented the two main options for STR. However, new strategies such as percutaneous intramyocardial septal radiofrequency ablation (PIMSRA), micro-coil septal embolization and transapical beating-heart septal myectomy (TA-BSM), have emerged as possible alternatives (Fig. 3). These techniques offer less invasive alternatives with promising efficacy. Compared to surgical myectomy, they present lower complication rates (5–10%), such as fewer conduction disturbances. They however tend to achieve a smaller reduction in LVOT gradient, especially in patients with complex anatomies [52]. Many of these new SRT techniques remain limited to specialized centres or are still in early-phase experience, with limited long-term data. Careful patient selection and individualized therapeutic planning remain essential. Moreover, MI and GT may promptly change LVOTO SRT indications.
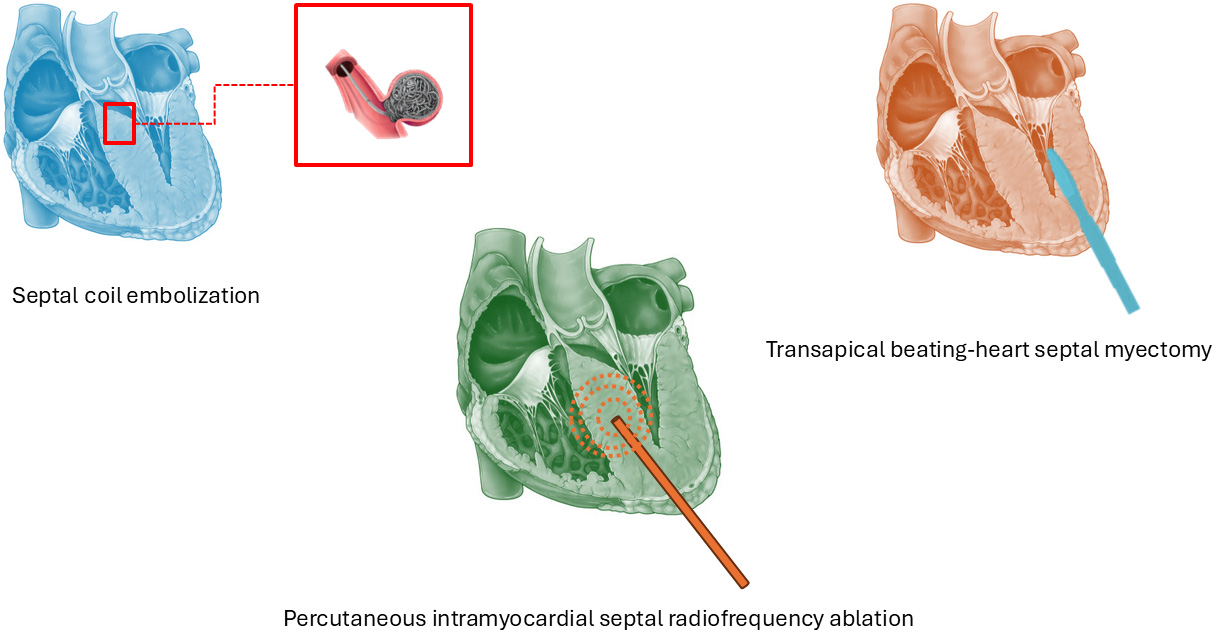 Fig. 3.
Fig. 3.
Emerging percutaneous and surgical techniques.
PIMSRA is a catheter-based SRT that delivers controlled radiofrequency energy directly into the hypertrophied septum via a specialized needle-electrode system with expandable arms. This creates a customizable, elliptical-shaped lesion through localized myocardial heating (up to 80 °C), while sparing the endocardial surface and protecting the conduction system [53]. The procedure is performed via transapical access under echocardiographic and CT guidance. Studies have shown significant reductions in LVOT gradient and improvements in NYHA functional class [54]. In a large cohort of 200 patients, resting gradient decreased from 79.2 to 14.0 mmHg at 12 months, with no need for permanent pacemaker implantation [55]. Subsequent studies confirmed a gradient reduction of over 80% and notable symptom relief. When compared with surgical myectomy, PIMSRA offers similar efficacy with substantially lower complication rates [56, 57].
Septal embolization with micro-coils offers a mechanical alternative to ASA. It also imitates local myocardial infarction by selectively occluding a septal artery, but leaving behind the possible toxicity of alcohol [58]. It is less likely to affect the conduction tissue, thus reducing the risk of permanent atrioventricular block and the need for permanent pacing [58]. A 58% reduction in LVOT gradient has been previously reported.
TA-BSM is a novel surgical procedure that allows direct septal resection through mini-thoracotomy and LV apex access. It is performed while the heart is beating (without cardiopulmonary bypass), and employs a retractable coring device guided by real-time transesophageal echocardiography [59]. Recent studies report high procedural success (91%), significant LVOT gradient reduction, and low complication rates, including no need for permanent pacing [59, 60, 61]. TA-BSM appears effective even in complex anatomical variants and shows comparable short- and mid-term outcomes to conventional Morrow surgery, with the added benefits of less invasiveness and real-time control. Long-term data are still pending.
Endocardial radiofrequency septal ablation offers precise endocardial targeting of the septal myocardium through guided electroanatomic mapping and intracardiac echocardiography. Short-term results have shown significant LVOT gradient reduction and symptomatic relief. However, comparative analyses suggest slightly lower efficacy than ASA and PIMSRA, in terms of septal thickness reduction [62, 63].
Robotic-assisted minimally invasive myectomy combines the precision of robotic platforms with reduced surgical trauma. Although still in the early stages of adoption, initial case series have demonstrated its feasibility and safety, particularly in elderly or high-risk patients [64].
High-intensity focused ultrasound employs extracorporeal energy delivery to induce precise myocardial necrosis. It has shown potential in canine models, presenting itself as an encouraging non-invasive alternative [65].
Transthoracic laser ablation (TTLA) involves percutaneous insertion of a laser fibre into the interventricular septum, delivering laser energy under magnetic resonance imaging or transesophageal echocardiogram guidance [66]. Its primary goal is to induce targeted myocardial coagulation necrosis, resulting in septal thinning and fibrosis. Preclinical animal studies have demonstrated feasibility and accuracy [67]. However, validation in humans is still required.
Multiple genes have been associated with HCM. According to the latest ClinGen Hereditary Cardiovascular Disorders Gene Curation Expert Panel, 29 genes have moderate, strong, or definitive evidence of association with HCM [68]. Most of these are sarcomeric genes, with MYBPC3 and MYH7 accounting for approximately 70–80% of genotype-positive cases [26].
Genetic testing in HCM carries a Class I recommendation according to ESC guidelines. Approximately 30–40% of patients with HCM will have a pathogenic or likely pathogenic variant identified [1]. A positive result has significant clinical implications, especially for initiating cascade screening in family members. While genotype status is not yet fully integrated into treatment algorithms, the development of GT is expected to change this in the future.
GT aims to correct the disease at the DNA level [69]. It may be applied through different mechanisms: gene replacement, gene editing and gene silencing. While gene replacement and editing are typically designed as one-time therapies, gene silencing requires recurrent administration to maintain therapeutic effect. Adeno-associated virus (AAV) vectors are commonly used for cardiac GT delivery due to their natural cardiotropism.
Numerous clinical trials employing different GT approaches have been conducted (Table 5).
| Gene therapy strategy | Mechanism of action | Main use | Limitations | Preclinical evidence | Clinical translation |
Gene Replacement
|
Delivery of a functional gene copy to restore normal protein expression. | Loss of function variants associated with haploinsufficiency. | - Dose dependent. | MYBPC3 knock-in mice: |
TN-201: AAV9-MYBPC3. Ongoing Phase 1 trial. |
Gene Editing |
Precise correction of pathogenic variants by insertion, deletion or conversion of nucleotides. | Gain of function mutations and missense mutations. | - Allele specificity critical. - Atrial editing suboptimal. - Off-target editing. |
MYBPC3 p.W1098X: correction of 3.65% of mutations; no phenotype. MYH7 p.R403Q: |
Not yet in clinical trials. |
Gene Silencing
|
siRNA-mediated selective degradation of mutant mRNA while preserving wild-type expression. | Dominant-negative mutations. | Requires heterozygosity with tolerable haploinsufficiency. Recurrent administration for long term efficacy. |
MYH6 p.R403Q: 80% mutant knockdown, no phenotype. | Not yet in clinical trials. |
mRNA, Messenger Ribonucleic Acid; siRNA, Small Interfering Ribonucleic Acid.
Gene replacement strategies targeting MYBPC3 have shown encouraging
results in both animal and human models. Initial approaches used trans-splicing
techniques, later evolving to full-length gene delivery. In a homozygous
MYBPC3 knock-in mouse model (c.722G
Gene editing approaches have also demonstrated therapeutic potential. One study
targeting a premature stop codon mutation (p.W1098X) in MYBPC3
achieved a correction rate of approximately 3.65% of total mutations at 6
months post-treatment. Despite the modest editing efficiency, no pathological
phenotype was observed during follow-up [74]. Further preclinical work using
CRISPR-Cas9 systems with adenine base editors in mouse models carrying the
MYH7 c.1208G
Finally, gene silencing has been successfully applied to target MYH6 mutations. In a heterozygous HCM mouse model carrying the R403Q mutation, administration of a mutation-specific siRNA (403i) resulted in an 80% reduction of the mutant transcript, while sparing approximately 80% of the wild-type allele. After 6 months of follow-up, treated mice showed no evidence of myocardial hypertrophy or fibrosis [77]. Importantly, this approach also demonstrated the ability to silence multiple mutations within the same gene by targeting shared single nucleotide polymorphisms, potentially broadening its applicability.
While GT shows promising preclinical results in HCM, several challenges limit its clinical application.
Firstly, AAV vectors can trigger immune responses leading to hepatotoxicity, myocarditis, and neurotoxicity. Dosing must be carefully managed to avoid adverse effects, and treatment is contraindicated in patients with pre-existing neutralizing antibodies [78, 79]. Packaging capacity of AAV is limited to ~4.7 kb, necessitating complex multi-vector delivery for large transgenes or editing tools [76]. Alternative vectors are under investigation [80].
Secondly, CRISPR designs must be allele-specific to avoid off-target effects. Normal gene function must be preserved and oncogenesis evaded. The need for mutation-specific sgRNAs also raises concerns about the scalability and cost-effectiveness of therapies for rare variants. Polygenic or non-Mendelian HCM forms may not benefit.
Clinical trial design is complicated by small sample sizes, ethical concerns due to incomplete penetrance and variable expressivity, and uncertainties about timing and disease reversibility. Evaluation often requires invasive biopsies, with surrogate endpoints being necessary. Long-term efficacy, durability of single-dose treatments, and optimal therapeutic windows remain undefined. To date, the longest published preclinical follow up lasted just 34 weeks [71].
Finally, access and affordability present major obstacles. High development costs, regulatory complexity, and limited availability may restrict treatment to high-resource settings. Regulatory hurdles are sure to arise due to GT complexity, safety concerns and long-term effects. Specific regulatory frameworks must be created, with a likely long approval process.
Despite these significant barriers, GT still represents a therapeutic revolution, sparking the hope that, in the future, gene-targeted approaches may offer curative potential for select subtypes of HCM [10].
During the last few years, a revolution in HCM has occurred; and a future revolution is to come. New SCD risk stratification methods incorporating technological advances and AI, are expected to emerge, improving ICD decision-making in HCM patients. As MI studies come to light, oHCM treatment algorithms may evolve, potentially positioning novel therapies as first-line options. Specific nHCM treatment is due to emerge. Septal ablation techniques will likely be reserved for non-responders, employing novel methods to improve outcomes and reduce complications. Finally, GT will soon become available for certain HCM genotypes enabling tailored, definitive treatments. Future prospects may even include preventive treatments for genetic carriers. HCM management will shift from phenotype-based to genotype-specific approaches, ushering us into the era of precision personalized medicine.
AAV, Adeno-Associated Viral Vectors; AI, Artificial Intelligence; ASA, Alcohol Septal Ablation; CMR, Cardiac Magnetic Resonance; CRISPR-Cas9, Clustered Regularly Interspaced Short Palindromic Repeats Associated Cas9 Nucleases; ECV, Extracellular Volume; EF, Ejection Fraction; GT, Gene Therapy; HCM, Hypertrophic Cardiomyopathy; ICD, Implantable Cardioverter-Defibrillators; KCCQ-CSS, Kansas City Cardiomyopathy Questionnaire–Clinical Summary Score; LAVI, Left Atrial Volume Index; LGE, Late Gadolinium Enhancement; LV, Left Ventricular; LVEF, Left Ventricular Ejection Fraction; LVMI, Left Ventricular Mass Index; LVOT, Left Ventricular Outflow Tract; LVOTO, Left Ventricular Outflow Tract Obstruction; MACE, Major Adverse Cardiovascular Event; MI, Myosin Inhibitors; mRNA, Messenger Ribonucleic Acid; nHCM, Non-Obstructive Hypertrophic Cardiomyopathy; oHCM, Obstructive Hypertrophic Cardiomyopathy; pVO2, Maximum Oxygen Consumption; PIMSRA, Percutaneous Intramyocardial Septal Radiofrequency Ablation; REMS, Risk Evaluation and Mitigation Strategy; SCD, Sudden Cardiac Death; sgRNA, single guide RNA; siRNA, small interfering RNA; SRT, Septal Reduction Therapy; TA-BSM, Transapical Beating-Heart Septal Myectomy; TTLA, Transthoracic Laser Ablation; VF, Ventricular Fibrillation; VT, Ventricular Tachycardia.
All authors meet the 4 criteria for authorship as outlined in the authorship policy. APA and JPD designed the research study. APA and JPD performed the research. APA, HLG, ALP, AVR and CJM wrote the manuscript. There was a conception and design meeting with all the authors. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
Julian Palomino-Doza is partially supported by the British Heart Foundation’s Big Beat Challenge award to CureHeart (BBC/F/21/220106).
The authors declare no conflict of interest.
Artificial intelligence has been partially used for the elaboration of some of the images of this paper. The authors reviewed and edited the content as needed and takes full responsibility for the content of the publication.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.