





 , Jiyao Xu 2,†, Chenxia Wu 1,3, Minyan Wang 4, Tingting Chen 5, Conghua Ji 4, Wei Mao 3,5,*
, Jiyao Xu 2,†, Chenxia Wu 1,3, Minyan Wang 4, Tingting Chen 5, Conghua Ji 4, Wei Mao 3,5,*
1 The First School of Clinical Medicine, Zhejiang Chinese Medical University, 310053 Hangzhou, Zhejiang, China
2 Department of Cardiology, Shanxi Cardiovascular Disease Hospital, 030027 Taiyuan, Shanxi, China
3 Cardiovascular Department, Zhejiang Hospital (Affiliated Zhejiang Hospital, Zhejiang University School of Medicine), 310030 Hangzhou, Zhejiang, China
4 School of Public Health, Zhejiang Chinese Medical University, 310053 Hangzhou, Zhejiang, China
5 Zhejiang Key Laboratory of Integrative Chinese and Western Medicine for Diagnosis and Treatment of Circulatory Diseases, Zhejiang Hospital, 310030 Hangzhou, Zhejiang, China
†These authors contributed equally.
Abstract
This article reviews the latest research progress (2018–2025) on the molecular mechanisms linking glucose and lipid metabolism disorders (GLMDs) to cardiovascular injury, specifically atherosclerotic cardiovascular disease (ASCVD), diabetic cardiomyopathy (DbCM), heart failure (HF), and cardiac autonomic neuropathy (CAN). This review employed a targeted analysis of key publications from the PubMed, Web of Science, and EMBASE databases, as well as citation tracking, prioritizing molecular pathways and interventions for these four complications. The key mechanisms include: metabolic inflammation: the advanced glycation end products (AGEs)–receptor of AGE (RAGE) axis activates NF-κB, promotes vascular cell adhesion molecule-1 (VCAM-1)/monocyte chemoattractant protein-1 (MCP-1) overexpression, and accelerates monocyte infiltration; myocardial lipotoxicity: CD36 mediates fatty acid overload → mitochondrial damage → cyclic guanosine monophosphate-adenylate synthetase (cGAS)-STING pathway activation → myocardial apoptosis; metabolic memory: hyperglycemia continuously releases small extracellular vesicle (sEV) miR-15-16 clusters through the O-GlcNAc–CaMKIIδ–STAT1 loop, mediating remote myocardial injury; gut–heart axis disorder: Trimethylamine N-Oxide (TMAO) promotes thrombosis and endothelial injury. Precision strategies based on the above mechanisms, such as SGLT2 inhibitors to improve myocardial energy metabolism, targeting acyl-coenzyme A binding protein (ACBP)/TGR5 to alleviate lipotoxicity, and microbiota regulation, have demonstrated potential in clinical research. Future focus should include (1) GLMD heterogeneity typing; (2) tissue-targeted delivery system; (3) multi-omics–AI dynamic risk modeling.
Keywords
- glucose and lipid metabolism disorders
- cardiovascular disease
- atherosclerosis
- molecular mechanisms
- precision medicine
- metabolic memory
- cGAS-STING
The global prevalence of diseases related to glucose and lipid metabolism disorders (GLMDs) (such as diabetes, metabolic fatty liver disease, and obesity) continues to rise [1], and cardiovascular complications caused by GLMDs have become the leading cause of death [2, 3]. The core pathological features of GLMDs include insulin resistance (IR) and ectopic lipid deposition [4], and their occurrence is the result of the combined effects of genetic susceptibility, environmental factors (such as diet and lack of exercise), and epigenetic regulation [5]. The key pathological processes involve insulin signal transduction disorders (such as IRS/PI3K/Akt pathway inhibition) [6], neuroendocrine regulation imbalance (such as hypothalamic-pituitary-adrenal axis (HPA axis) activation, increased sympathetic nerve tone) [3], redox homeostasis disruption (excessive production of reactive oxygen species (ROS)) [7], persistent low-grade inflammatory response [8], and intestinal microecological disturbances [2, 9, 10, 11]. These processes interact with each other to form a vicious cycle, ultimately leading to vascular endothelial damage, myocardial remodeling and abnormal neural regulation, significantly increasing the risk of atherosclerotic cardiovascular disease (ASCVD), heart failure (HF) and cardiac autonomic neuropathy (CAN) [12]. Studies have shown that long-term exposure to high blood sugar and high triglyceride environments significantly increases the risk of cardiovascular events such as myocardial infarction and stroke [13, 14]. An elevated triglyceride-glucose index (TyG index) is significantly associated with an increased risk of cardiovascular events [15, 16]. It is worth noting that although traditional risk factor management (such as glucose lowering and lipid regulation) can partially reduce the risk of cardiovascular disease (CVD), a large number of patients (especially those with good blood sugar control) still have high residual risks [17, 18], highlighting the urgency of in-depth exploration of the deep molecular mechanisms of GLMD-induced cardiovascular damage and the development of precise intervention strategies. The advancement of multi-omics technologies (genomics, transcriptomics, proteomics, metabolomics, microbiome) provides powerful tools for systematic analysis of these mechanisms [9, 19].
Traditional risk management has a limited effect on the residual cardiovascular risk of GLMD patients, highlighting the urgency of exploring deep molecular mechanisms and developing precision interventions. This article aims to review the progress of the molecular mechanisms of GLMD-related cardiovascular damage (ASCVD/diabetic cardiomyopathy (DbCM)/CAN), with a special focus on emerging pathways such as metabolic memory, lipotoxicity and microbiota-host dialogue, and evaluate the clinical translation potential of targeted intervention strategies.
Chronic low-grade inflammation (metabolic inflammation, Metaflammation) is the core link between GLMD and ASCVD [20]. Hyperglycemia, elevated free fatty acids (FFA) and advanced glycation end products (AGEs) jointly activate the inflammatory pathway of endothelial cells (ECs), destroying the barrier function and promoting monocyte adhesion and infiltration and foam cell formation.
Sustained hyperglycemia leads to the massive generation of AGEs. AGEs bind to
the ECs surface receptor RAGE, triggering a strong activation of the downstream
NF-
Lipoprotein(a) [Lp(a)]: Lp(a) has the “dual identity” of promoting
atherosclerosis and thrombosis. Its apolipoprotein(a) [apo(a)] structure is
highly homologous to plasminogen and can competitively inhibit plasminogen
activation, weakening fibrinolytic activity [24, 25]. A Multinational prospective
cohort study showed that high Lp(a) level (
In the GLMD state, HDL undergoes oxidation, inflammation and other modifications, such as nitration of apolipoprotein A-I and accumulation of serum amyloid A, leading to impaired cholesterol reverse transport (RCT) ability (ATP binding cassette transporter A1/G1 pathway disorder); The anti-inflammatory and antioxidant functions significantly decreased (inactivation of paraoxonase 1 and increase in pro oxidative LDL); Loss of endothelial protective function (reduced eNOS activation and insufficient NO production) [27]. Studies have shown that the cholesterol reverse transport capacity of HDL in diabetes patients is controversial, but its antioxidant function is clearly impaired [28, 29].
EAT is a fat pad that is close to the myocardium and coronary arteries. It
undergoes significant pathological changes under the GLMD state, manifested by
adipocyte hypertrophy, macrophage infiltration (increase in proinflammatory M1
type) [30], increased secretion of proinflammatory factors (such as
TNF-
These synergistic interactions are schematically summarized in Fig. 1.
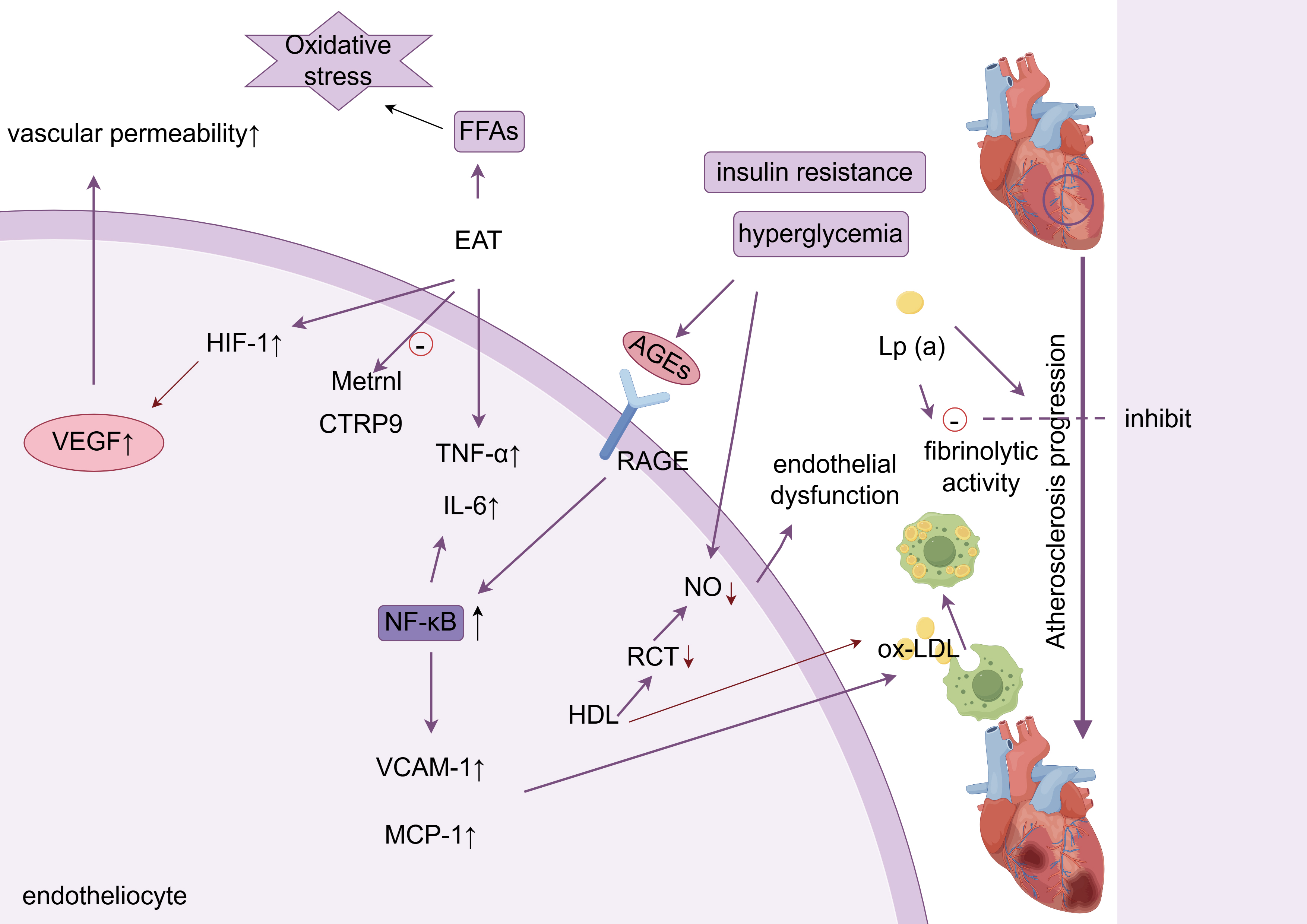 Fig. 1.
Fig. 1.
Metabolic inflammation and endothelial dysfunction. ox-LDL,
oxidized low-density lipoprotein; AGEs, advanced glycation end products; VEGF,
vascular endothelial growth factor; RCT, cholesterol reverse transport; RAGE,
receptor for advanced glycation endproducts; NO, nitric oxide; FFA, free fatty
acids; HIF-1, hypoxia-inducible factor-1; EAT, epicardial adipose tissue; TNF-
GLMD (especially diabetes) leads to a pathological change in the myocardial energy metabolism pattern, which is characterized by excessive reliance on fatty acid oxidation (FAO) for energy supply, impaired glucose utilization, mitochondrial dysfunction and accumulation of toxic lipid intermediates, collectively referred to as “lipotoxicity”, which is the core mechanism of DbCM [34, 35].
In physiological conditions, the heart has metabolic flexibility and can switch between glucose and fatty acid oxidation according to substrate supply. In GLMD, impaired myocardial insulin signaling leads to reduced translocation of glucose transporter 4 (GLUT4) to the cell membrane, and myocardial glucose uptake and utilization are significantly reduced [36, 37]. As compensation, myocardial cells increase fatty acid uptake and oxidation. Fatty acid transporter CD36 plays a key role in this process. Studies have revealed the regulation of CD36 by the bile acid-TGR5 pathway: In the diabetic state, the level of bile acids (especially deoxycholic acid DCA) that activate TGR5 in plasma is reduced. TGR5 is a G protein-coupled bile acid receptor expressed in cardiomyocytes. Absence of TGR5 signaling (e.g., cardiomyocyte-specific Tgr5 knockout mice) or attenuation (diabetic state) promotes palmitoylation of CD36 protein by upregulating the expression and activity of palmitoyltransferase DHHC4. Enhanced palmitoylation of CD36 increases its translocation to the cell membrane, significantly increasing fatty acid uptake by cardiomyocytes. This CD36 Uncontrolled membrane translocation is an important mechanism of myocardial lipid overload [34].
The influx of excess fatty acids exceeds the mitochondrial
Autophagy is a key process for clearing damaged organelles (such as dysfunctional mitochondria) and misfolded proteins. In diabetic myocardium, autophagic flux is often impaired, manifested by decreased expression of key autophagic proteins (such as Beclin-1, microtubule-associated protein 1 light chain 3-II (LC3-II)) or impaired fusion of autophagosomes with lysosomes [42].
Severe or persistent mitochondrial damage can lead to the release of
mitochondrial DNA (mtDNA) into the cytoplasm. The mtDNA in the cytoplasm is
recognized by the DNA sensor cyclic guanosine monophosphate-adenylate synthetase
(cGAS), which activates the adaptor protein stimulator of interferon genes
(STING). Activated STING then recruits and activates TANK
binding kinase 1 (TBK1), phosphorylates the transcription factor interferon
regulatory factor 3 (IRF3), and ultimately induces the production of
proinflammatory cytokines such as type I interferon (such as IFN-
High glucose and lipotoxic environment enhance cGAS-STING
activation: Enhanced mitochondrial damage: A hyperglycemic environment increases
ROS production, leading to mitochondrial dysfunction and oxidative damage to
mtDNA, and increasing mtDNA leakage [45]. Lipotoxicity further exacerbates
mitochondrial damage. The cGAS-STING pathway drives the myocardial
inflammatory cascade through the TBK1-IRF3-IFN
acyl-coenzyme A binding protein (ACBP) and myocardial lipotoxicity: In diabetes cardiomyopathy, ACBP aggravates myocardial lipotoxicity by regulating fatty acid metabolism and affecting myosin function. ACBP disrupts the contractile structure by binding to the sarcomere protein MyBPC3. The upregulation of ACBP leads to disturbances in fatty acid metabolism, further triggering oxidative stress and mitochondrial damage, ultimately resulting in myocardial energy metabolism remodeling and apoptosis. Myocardial specific knockout of ACBP can improve cardiac function in diabetes mice, suggesting that ACBP is a potential double effect target [46].
Lipid droplet dynamic imbalance and ceramide toxicity: When the fatty acid uptake rate far exceeds the oxidation capacity, neutral triglycerides (TG) are stored in myocardial cells in the form of lipid droplets. Moderate lipid droplet formation has a protective effect in buffering lipotoxicity, but this effect is limited. In severe GLMD, myocardial lipid droplets are excessively deposited and lipid droplet mobilization and utilization are blocked. In addition, saturated fatty acids (such as palmitic acid) can be converted into ceramide, a highly cytotoxic sphingolipid. Ceramide promotes macrophage inflammatory response and plaque instability by activating specific receptors (such as cysteine leukotriene receptor 2 CYSLTR2, purinergic receptor P2RY6), exacerbating atherosclerosis [47]. In addition, ceramide further exacerbates metabolic disorders by inhibiting the insulin signaling pathway and mitochondrial metabolism. AMP-activated protein kinase (AMPK) is a key kinase that regulates energy metabolism. Its activation (such as by drugs or exercise) can promote FAO and significantly reduce ceramide accumulation, and is an important target for improving lipotoxicity [48].
In summary, lipid toxicity is mediated by CD36 mediated fatty acid overload, mitochondrial ROS burst, and activation of the cGAS STING pathway, forming a vicious cycle of ‘lipid oxidative stress inflammation’, which is the core driving mechanism of DbCM. Recent studies have further revealed the interaction between mitochondrial quality control imbalance and lipid toxicity, suggesting that the mitochondrial deacetylase SIRT4 may participate in lipid toxicity processes by regulating CD36 palmitoylation and mitochondrial autophagy, providing a new perspective for targeting mitochondrial lipid metabolism interactions.
The multi-level metabolic alterations are illustrated in Fig. 2.
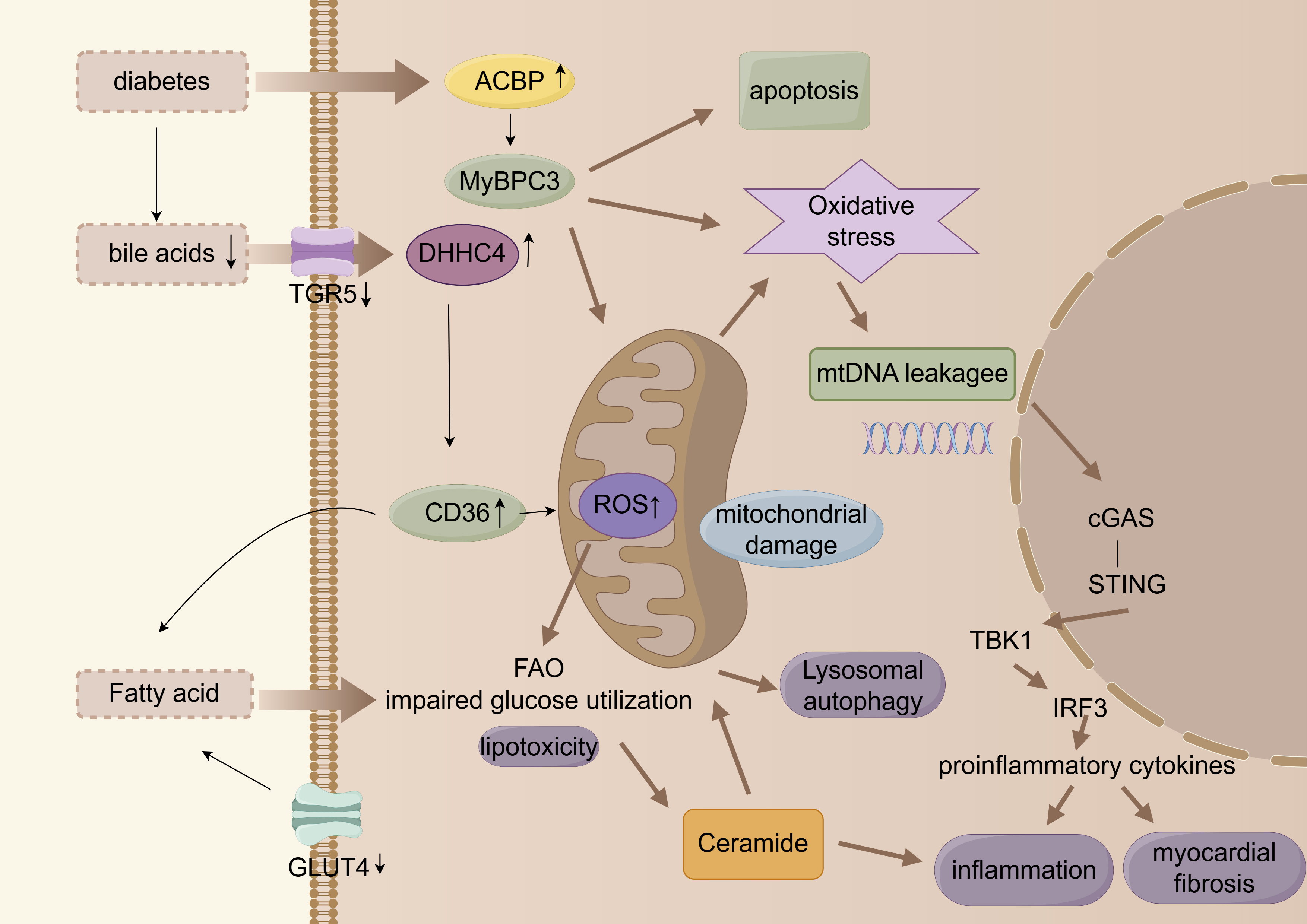 Fig. 2.
Fig. 2.
Myocardial energy metabolic remodeling. cGAS, cyclic guanosine
monophosphate-adenylate synthetase; FAO, fatty acid oxidation; ACBP, acyl
coenzyme A binding protein. Arrows indicate the direction of change in the glucolipid metabolic disorder (GLMD) state: up arrows (
Clinical observations (such as the Diabetes Control and Complications Trial/Epidemiology of Diabetes Intervention and Complications Study (DCCT/EDIC))
have confirmed that even if blood sugar is well controlled in the later stage,
previous exposure to high blood sugar can still lead to the continued progression
of cardiovascular complications of diabetes. This phenomenon is called
“metabolic memory” or “hyperglycemia memory” (Hyperglycemic Memory, HGM) [49, 50]. Its molecular mechanism involves multi pathway synergistic effects: O-GlcNAc
glycosylation/CaMK2a positive feedback loop: Hyperglycemia increases protein
O-linked N-acetylglucosamine via the hexosamine biosynthesis pathway (HBP)
(O-GlcNAc) modification. O-GlcNAc modification specifically activates the
calcium/calmodulin-dependent protein kinase II
Sustained release of the sEV miR-15-16 cluster: Activated
CaMKII
Forkhead box protein O1 (FoxO1)-mediated cardiomyocyte apoptosis: Endothelial-derived sEVs enriched in the miR-15-16 cluster are taken up by cardiomyocytes, and miR-15-16 targets, and suppresses Serum/Glucocorticoid Regulated Kinase 1 (SGK1), Proto-OncogeneSerine/Threonine Kinase (RAF1), and Vascular Endothelial Growth Factor A (VEGFA), reducing Akt phosphorylation (p-Akt) and thereby decreasing inhibition on the transcription factor FoxO1. This results in significantly increased FoxO1 expression, ultimately inducing cardiomyocyte apoptosis and cardiac dysfunction [51].
Clinical verification and significance: In the plasma sEV of diabetes patients with heart failure, the level of miR-15-16 was significantly increased, and was significantly negative with left ventricular ejection fraction (LVEF). This indicates that sEV miR-15-16 is an independent biomarker of cardiac dysfunction independent of current blood glucose status and a key mediator of metabolic memory induced sustained myocardial injury. This study emphasizes the importance of early reinforcement of glucose control or inhibition of O-GlcNAc modification in blocking metabolic memory circuits. The metabolic memory of hyperglycemia is illustrated in Fig. 3.
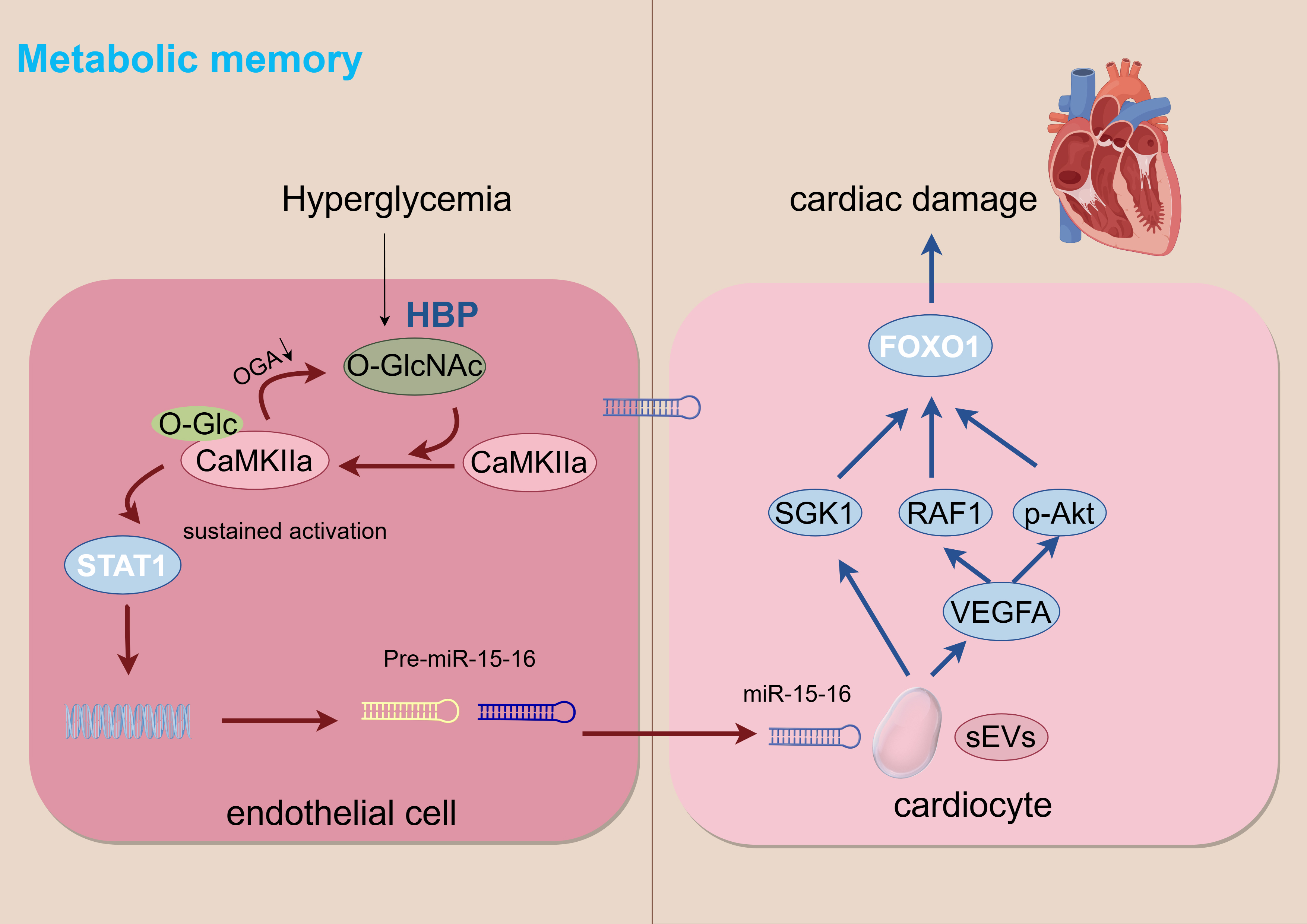 Fig. 3.
Fig. 3.
Metabolic memory. HBP, hexosamine biosynthesis pathway. Arrows indicate the direction of change in the glucolipid metabolic disorder (GLMD) state: down arrows (
CAN is a serious complication of GLMD, characterized by sympathetic/parasympathetic nerve imbalance, manifested by decreased heart rate variability (HRV), resting tachycardia, postural hypotension and painless myocardial ischemia, significantly increasing the risk of malignant arrhythmias and sudden cardiac death. Its pathogenesis is closely related to multiple factors such as chronic hyperglycemia-induced oxidative stress, visceral fat inflammation and autoimmune attack. Early identification of CAN requires the combination of functional imaging and dynamic biomarker detection [53, 54].
HRV analysis through a 24-hour dynamic electrocardiogram is the gold standard for diagnosing CAN. The low frequency/high frequency power ratio (LF/HF) of diabetic patients is significantly higher than that of healthy people, which clearly indicates that the sympathetic nerve tone is dominant over the parasympathetic nerve [53, 55].
Visceral adipose tissue (VAT) is an important source of inflammation and FFA.
Under GLMD, VAT releases a large amount of FFA and proinflammatory factors (such
as TNF-
The prevalence of CAN is significantly higher in patients with autoimmune diabetes (such as latent autoimmune diabetes in adults LADA and classic type 1 diabetes). A cross-sectional study showed that the prevalence of CAN in patients with positive anti-islet cell antibodies (ICA) was significantly higher than that in the ICA-negative group [57].
Gut microbiota and its metabolites constitute a complex “gut-heart axis” that plays an increasingly clear role in GLMD-related cardiovascular damage [11]. These metabolites affect cardiovascular health through multiple mechanisms, including regulation of inflammatory responses, lipid metabolism, and insulin sensitivity.
Harmful Metabolites:
Trimethylamine oxide (TMAO): Gut microbiota metabolizes choline and carnitine in food to produce trimethylamine (TMA), which is oxidized to TMAO by the liver enzyme flavin monooxygenase 3 (FMO3). High TMAO levels are independently associated with increased risk of ASCVD and HF. TMAO promotes atherosclerosis by promoting foam cell formation, enhancing platelet reactivity (increasing thrombotic risk), and inducing endothelial dysfunction [58, 59].
GLMD is often accompanied by changes in intestinal flora, which affects bile acid metabolism. The abnormal increase in the proportion of visceral fat (such as deoxycholic acid DCA) may promote inflammation or affect farnesoid X receptor (FXR)/TGR5 signaling is involved in cardiovascular damage [34, 60].
Short chain fatty acids (SCFAs): SCFAs (such as acetate, propionate, and butyrate) are produced by fermentation of dietary fiber through gut microbiota. They have anti-inflammatory properties, maintaining intestinal barrier integrity, and improving insulin sensitivity [61]. GLMD status is often accompanied by a decrease in the abundance of SCFA producing bacteria (such as Faecalibacterium prausnitzii and Roseburia), a decrease in SCFA levels, and a weakening of their cardiovascular protective effects [62, 63].
Intestinal flora disorder (dysbiosis) is often accompanied by impaired
intestinal barrier function (“leaky gut”), which leads to the translocation of
bacterial endotoxin LPS into the blood circulation. LPS and FFA synergistically
promote macrophage M1 polarization and drive TNF-
The gut-brain-heart axis dysregulation is visualized in Fig. 4.
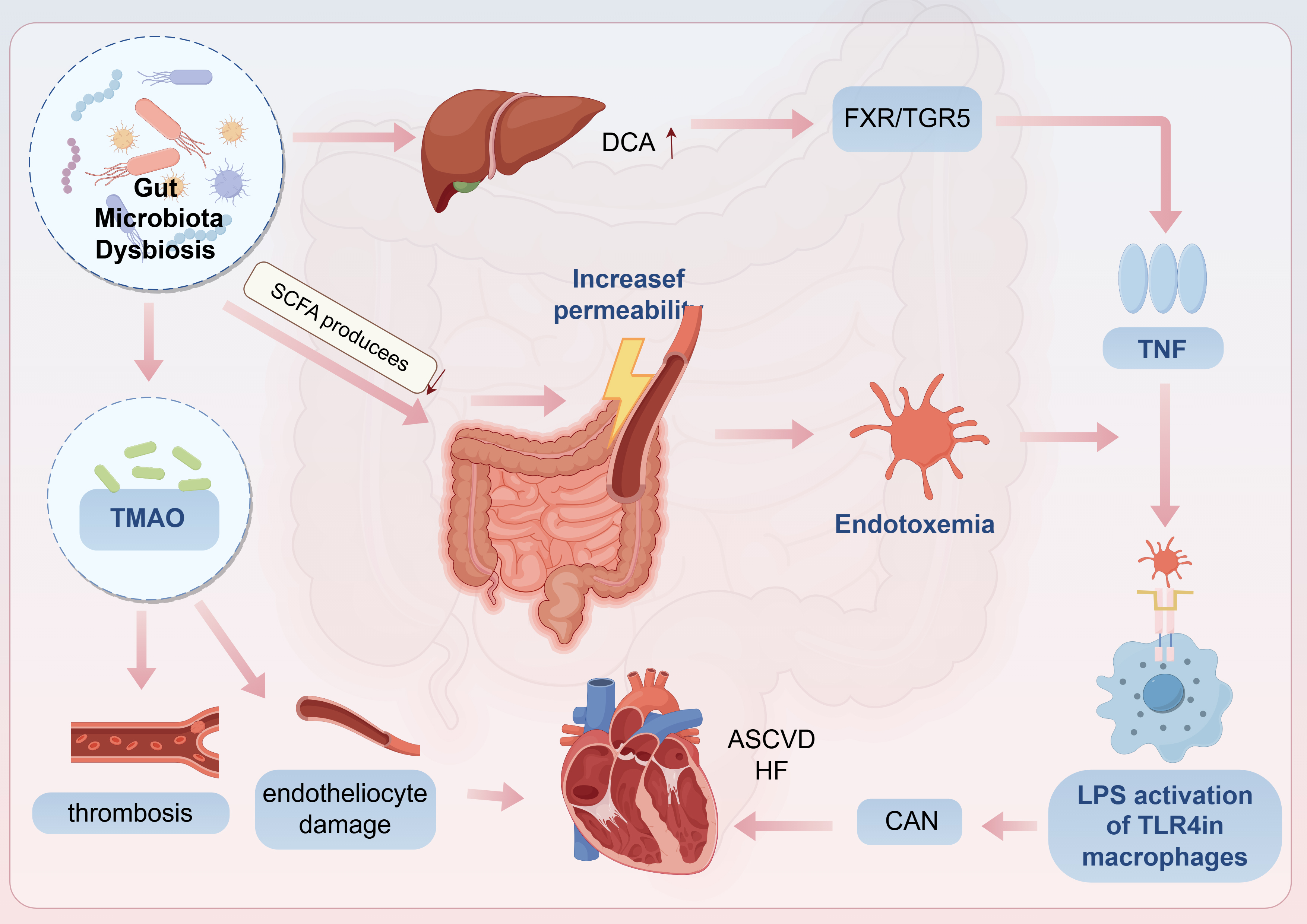 Fig. 4.
Fig. 4.
CAN and Gut microbiota dysbiosis. CAN, cardiac autonomic
neuropathy; DCA, deoxycholic acid; TNF, tumor necrosis factor; HF, heart failure;
ASCVD, atherosclerotic cardiovascular disease. Arrows indicate the direction of change in the glucolipid metabolic disorder (GLMD) state: up arrows (
The association between GLMD and cardiovascular events exhibits a significant
dose-response relationship and risk amplification. The risk of cardiovascular
events in patients with diabetes is 2–4 times (male) to 2–7 times (female)
higher than that in non-diabetic patients, and the absolute risk increases
significantly with the younger age of diabetes (onset
Dyslipidemia and myocardial infarction: diabetes with dyslipidemia (characterized by high triglycerides and low high-density lipoprotein cholesterol (HDL-C)) significantly increases the risk of coronary heart disease, in which the elevated level of low-density lipoprotein cholesterol (LDL-C) is an independent risk factor for severe coronary heart disease (OR = 1.151, p = 0.046) [65]. Lipid metabolism disorder has existed in pre diabetes and is closely related to the accumulation of ectopic fat [64].
The pathological impact of visceral fat is not limited to the release of inflammatory factors. Clinical imaging data show that obese T2DM patients have significantly increased EAT, and the increase in EAT is associated with a significant increase in coronary artery calcification score (CAC) [67, 68]. Its mechanism is closely related to the infiltration of proinflammatory factors (such as IL-6 and C-reactive protein CRP) secreted by EAT into the coronary artery, aggravating local inflammation and plaque instability [68].
These findings not only elucidate the strength of the association between GLMD and cardiovascular events but also provide critical insights for prevention and therapeutic strategies.
The latest progress in precision medicine for diabetes provides an important reference for precise intervention of GLMD-related cardiovascular damage. By integrating individual biological, environmental and lifestyle information, multi-omics technologies (such as genomics, transcriptomics, proteomics, and metabolomics) can be used to more accurately identify patient subtypes and provide precise targets for the prevention and treatment of cardiovascular complications.
The mechanism of SGLT2i is to inhibit sodium-glucose cotransporter 2 (SGLT2) in the proximal tubule of the kidney and to promote urinary glucose excretion [69]. SGLT2i confers cardiovascular protection that is partly independent of glucose lowering. Their pleiotropic effects include: (1) enhancing myocardial energy efficiency by shifting substrate utilization toward ketone bodies and alleviating lipotoxicity [70]; (2) improving sodium balance and hemodynamics through natriuresis and diuresis, thereby reducing cardiac preload and afterload [69]; and (3) decreasing ventricular stiffness by attenuating coronary inflammation- achieved via reductions in EAT volume and suppression of its pro-inflammatory secretome [71]. These agents are indicated for patients with type 2 diabetes mellitus who have established ASCVD or are at high risk for ASCVD, for those with HF with reduced ejection fraction (HFrEF) or preserved ejection fraction (HFpEF), and for individuals with chronic kidney disease (CKD). Applicable populations: T2DM patients with ASCVD or high risk of ASCVD, HF (with reduced ejection fractionHFrEF/with preserved ejection fractionHFpEF ), and CKD [69]. Those with elevated N-terminal pro-B-type natriuretic peptide (NT-proBNP) may especially benefit from these agents [72, 73].
GLP-1RAs work by activating GLP-1 receptors, thereby enhancing glucose-dependent insulin secretion, suppressing glucagon release, slowing gastric emptying, and increasing satiety. Their cardiovascular benefits are chiefly manifested by markedly lowering the risk of ASCVD events such as myocardial infarction and stroke-through anti-atherogenic actions that attenuate vascular inflammation, improve endothelial function, and stabilize plaques by shrinking the lipid-rich necrotic core and thickening the fibrous cap. The mechanism of GLP-1RA is to activate GLP-1 receptors to enhance glucose-dependent insulin secretion, inhibit glucagon, delay gastric emptying, and increase satiety. Cardiovascular benefits are mainly reflected in a significant decrease in the risk of ASCVD events (myocardial infarction, stroke): Anti-atherosclerosis benefits: Reduces vascular inflammation, improves endothelial function,and stabilizes plaques (reduces lipid necrotic core in plaques and increases fibrous cap thickness) [74].
GRK-biased adrenergic agonists are a new class of drugs that bias G protein coupling. The GRK2 pathway of the receptor (GPCR) reduces cAMP production, thereby reducing cardiac side effects and effectively promoting muscle glucose uptake. This drug has shown good efficacy and safety in the treatment of type 2 diabetes and obesity, and is expected to become a new treatment option for cardiovascular complications of diabetes [75].
TGR5 agonists: Activation of myocardial TGR5 signaling can inhibit DHHC4-mediated CD36 palmitoylation and membrane translocation, reduce myocardial fatty acid uptake, improve lipotoxicity and cardiac function, and provide a theoretical basis for the development of selective TGR5 agonists to treat DbCM [34].
Targeted ACBP (acyl coenzyme A binding protein) intervention: the expression of ACBP in diabetic myocardium is up-regulated. ACBP not only exacerbates the disorder of fatty acid metabolism, but also disrupts muscle filament structure and reduces contractility by abnormally binding to the sarcomere protein Myosin Binding Protein C (MyBPC3). Myocardial specific knockout of ACBP can improve cardiac function in diabetic mice, suggesting that ACBP is a potential double effect target [46].
The cGAS-STING pathway is a “common inflammatory hub” for multi-organ complications of diabetes (such as cardiomyopathy, nephropathy, and retinopathy), and its inhibitors show potential for precision intervention [45].
C-176 (STING dimerization inhibitor): significantly reduced myocardial
IFN-
H-151 (STING covalent inhibitor): In the myocardial infarction (MI) model (non-diabetic cardiomyopathy model), by specifically inhibiting the cGAS-STING-IRF3 pathway in infiltrating macrophages, it significantly reduced the type I interferon response, thereby: improving myocardial contractile function, and reducing cardiac fibrosis [76].
NT-proBNP is a marker of myocardial wall stress and is closely related to the occurrence and prognosis of HF. Incorporating NT-proBNP into cardiovascular risk assessment models (such as the Systemic Coronary Risk Assessment 2 (SCORE2) model) can significantly improve risk prediction capabilities, especially in identifying high-risk populations [77].
Plasma levels of the miR-15-16 cluster in small extracellular vesicles (sEVs), and are closely associated with diabetes-related metabolic memory and the risk of cardiac damage. These plasma sEV miR-15-16 cluster levels demonstrate a significant correlation with cardiac dysfunction in diabetic patients-independent of blood glucose or HbA1c levels-and represent a promising novel biomarker for assessing the risk of metabolic memory-associated cardiac injury [51].
Machine learning enables identification of high-risk individuals for cardiovascular events among diabetic patients through analysis of extensive clinical datasets. By integrating clinical, laboratory, and imaging data within ML algorithms, accurate predictive models can be developed to enhance cardiovascular risk stratification [78]. Deep neural networks (DNNs) have shown significant promise in this domain: one study successfully predicted 1-year all-cause mortality using only 12-lead electrocardiogram (ECG) voltage data via a DNN architecture, demonstrating robust performance across large-scale validation [79] . This DNN-based approach not only improves prediction accuracy but also offers a powerful tool for early cardiovascular disease screening and intervention.
Microbial regulation: Dietary intervention (such as the Mediterranean diet): The Mediterranean diet, which is rich in fiber, polyphenols, and unsaturated fatty acids, can promote the growth of beneficial bacteria (SCFA-producing bacteria), increase SCFA levels, and reduce serum inflammatory markers (such as high-sensitivity C-reactive protein hs-CRP) [80, 81]. Its cardiovascular protective effect is partly achieved by improving the microbiota.
Probiotics/prebiotics/synbiotics: Specific strains (such as certain bifidobacteria, lactobacilli) or prebiotics (such as inulin, oligofructose) have shown the potential to improve lipid metabolism, reduce TMAO, and reduce inflammation in animal and some human studies [82, 83].
Selective inhibition of harmful pathways: The development of drugs that inhibit bacterial TMA cleavage enzymes or host FMO3 activity to reduce TMAO levels is an active research direction [84]. Breakthroughs have been made in the clinical transformation of targeted TMAO, and early clinical studies of FMO3 inhibitors have shown potential [85], but long-term efficacy requires phase III trials to verify.
Metabolic memory intervention: Early intensive blood sugar control is the key
time window for blocking the formation of the
O-GlcNAc/CaMKII
Application of multi-omics technology in precision intervention: Multi-omics technology (including genomics, transcriptomics, proteomics and metabolomics) provides new tools for precision intervention of cardiovascular diseases. By integrating multi-omics data, the molecular network of disease progression can be revealed, new biomarkers and therapeutic targets can be identified, and personalized treatment strategies can be achieved [86].
To consolidate recent breakthroughs and translational needs, Table 1 (Ref. [26, 27, 42, 45, 46, 51, 56, 57, 58, 59, 71, 75, 77]) summarizes key advances and critical research gaps for the four primary cardiovascular complications of GLMD.
| Disease | Key advances | Identified research gaps |
| ASCVD | Role of Lp(a) in thrombosis (Wong et al. 2024) [26] | Lipoprotein dysfunction (Lp(a) thrombosis + HDL impairment) drives plaque instability |
| HDL dysfunction promotes plaque instability (Madaudo et al. 2024) [27] | Long-term effects of EAT modulation on plaque stability | |
| SGLT2i reduces EAT inflammation (Bao et al. 2025) [71] | ||
| DbCM | Mitochondrial autophagy impairment amplifies lipotoxicity (Dewanjee et al. 2021) [42] | Tissue-specific modulation of mitochondrial quality control |
| cGAS-STING pathway links lipotoxicity to inflammation (He et al. 2024) [45] | Safe delivery of STING inhibitors to cardiomyocytes | |
| ACBP disrupts sarcomere structure and exacerbates lipotoxicity (Wu et al. 2025) [46] | Clinical validation of ACBP inhibitors | |
| HF | sEV miR-15-16 as metabolic memory biomarker (Ding et al. 2025) [51] | Dynamic monitoring of sEV biomarkers |
| GRK-biased agonists improve myocardial glucose uptake (Motso et al. 2025) [75] | Pharmacokinetic optimization of GRK agonists for cardiac specificity | |
| NT-proBNP enhances cardiovascular risk prediction (Lehmacher et al. 2022) [77] | AI-integrated risk prediction for HF phenotypes | |
| CAN | FFA and cytokines activate neuronal TLR4-NF-κB signaling (Meng et al. 2022) [56] | Early biomarkers for subclinical CAN |
| Autoimmune diabetes increases CAN susceptibility (Risi et al. 2025) [57] | Neuron-targeted delivery of anti-inflammatory agents | |
| Gut microbiota metabolites (e.g., TMAO) drive endothelial injury and autonomic imbalance (Wen et al. 2022 [58], Tanase et al. 2020 [59]) | Clinical trials of microbiota-directed interventions |
DbCM, diabetic cardiomyopathy; HDL, high-density lipoprotein; ACBP, acyl-coenzyme A binding protein.
Define high-risk GLMD subtypes through integrated multi-omics (genomics, metabolomics, microbiome) and deep phenotyping (imaging, dynamic glucose monitoring). Key subtypes include: nflammation-dominant (elevated CRP/IL-6, EAT thickening); Severe lipotoxicity (myocardial lipid deposition, ACBP/CD36 overexpression); Metabolic memory-sensitive (persistently high sEV miR-15-16 levels); Dysbiosis-driven (reduced SCFA-producing bacteria, elevated TMAO-generating microbiota).
Many promising therapeutic targets (such as CD36 and STING within cardiomyocytes, and CaMKIIa within endothelial cells) are located in specific organelles or cell types. Systemic drug administration often faces challenges including off-target effects, dose-limiting toxicity, or difficulties in achieving effective concentrations at the target site. Developing heart/vascular-specific targeted nanocarriers (e.g., liposomes, engineered exosome carriers) or adeno- associated virus (AAV)-based gene therapy vectors is crucial for enhancing their efficacy and reducing side effects. For example, overcoming the myocardial-specific delivery bottleneck is essential for cGAS-STING targeted therapy: Existing inhibitors (e.g., C-176) administered systemically readily interfere with immune surveillance functions, potentially increasing the risk of infection. Solutions include developing myocardial-targeted liposomal encapsulation technology that utilizes highly expressed proteins in cardiomyocytes (such as cardiac troponin I (cTnI) antibody modification) to achieve drug enrichment.
Moving beyond single or a few biomarkers, we integrate static (genetic, epigenetic) and dynamic (transcriptomic, proteomic, metabolomic, microbiome, radiomic, continuous glucose/physiological monitoring) data streams. Machine learning and deep learning are leveraged to construct dynamically updatable, individualized cardiovascular risk prediction models. For instance: Integrating ECG-AI features (derived from electrocardiogram-artificial intelligence analysis), metabolic memory markers (e.g., sEV miR-15-16 clusters), radiomics (EAT volume), and continuous glucose monitoring (CGM) data enables the development of significantly more accurate dynamic risk stratification models.
GLMD drives multi-organ cardiovascular injury—encompassing ASCVD, DbCM, HF,
and CAN—through interconnected molecular cascades: Vascular inflammation via
AGEs-RAGE/NF-
LW & JX: Conceptualized the study, wrote the original draft, and created the visualizations. CW: Performed the formal statistical analysis and data curation and contributed to the writing of the methodology and results sections of the manuscript. MW: Contributed to methodology and validation of the experimental results, and critically reviewed the manuscript for important intellectual content, particularly on the methodological and analytical aspects. TC: Conducted the investigation and data collection, and participated in reviewing and editing the manuscript. CJ: Contributed to conceptualization, supervised the research, and reviewed & edited the manuscript. WM: Supervised the research, acquired funding, contributed to the study design and data interpretation, and reviewed & edited the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
The authors thank the funding bodies and the anonymous reviewers for their constructive feedback.
Supported by the National Key Research and Development Program of China (Grant No. 2023YFC3606201).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.