
- Academic Editor
Takayasu's arteritis (TAK) is a rare chronic arteritis that can lead to serious consequences. Understanding of the pathogenesis of TAK remains limited, and effective therapeutic strategies for this condition are lacking. Previous studies have suggested that there may be an association between TAK and the interaction between Mycobacterium tuberculosis infection and genetic susceptibility. Emerging data indicate that the nucleotide-binding and oligomerization domain (NOD)-like receptor pyrin domain-containing protein 3 receptor (NLRP3) inflammasome may be involved in the pathogenesis of TAK, potentially contributing to the initiation of the disease. This review summarizes the current epidemiological data, possible mechanisms, and targeting strategies of TAK, focusing on the involvement of the NLRP3 inflammasome in the pathogenesis of TAK, and provides new insights into the prevention and treatment of this condition.
Takayasu’s arteritis (TAK) is a chronic inflammatory disease that primarily
affects the aorta and its major branches, causing granulomatous inflammation and
fibrosis of the vessel wall. It is a rare disease, but can lead to severe
complications. TAK can affect large and medium arteries throughout the body,
causing various non-specific complications such as visual impairment, stroke,
myocardial infarction, heart failure, hypertension, and intermittent
claudication. Laboratory tests, while indicating inflammation with elevated
markers, are not specific to TAK, making diagnosis heavily reliant on angiography
and other radiological techniques [1]. The non-specific symptoms and lack of
efficient diagnostic methods, coupled with lower awareness among some clinicians,
contribute to its underdiagnosis and misdiagnosis, resulting in a mortality of
2–11% at 5 years and 14% at 15 years. In addition, 3–13% of patients require
interventional therapy to alleviate severe limb impairments [2]. Furthermore, TAK
creates a substantial economic burden, particularly in terms of increased
inpatient costs. A study found that patients with TAK had a mean additional
Despite decades since its discovery in the 1950s, the precise mechanisms of TAK remain extremely limited. The prevailing view suggests that TAK is an autoimmune disease where an unknown trigger initiates an immune response, leading to inflammation and the damage of arteries, tissues, and cells and causing the chemotaxis of macrophages and proliferation of fibroblasts and granulomas formation in the blood vessel walls. This leads to vascular stenosis, dilation, and even occlusion in large and medium-sized arteries, resulting in corresponding clinical symptoms [4].
Available data have shown some connection between factors and TAK, such as Mycobacterium tuberculosis infection or genetic susceptibility; however, the exact predisposing or detailed etiological factors are controversial, which makes the prevention and control of the disease a huge challenge. According to population surveys, the incidence of TAK in Asia shows an increasing trend [5]. It is estimated that in 2017, there were 5320 cases in Japan (95% CI: 4810–5820), whereas in 1994, this number did not exceed 5000 [6, 7]. Treatment involves both surgical and pharmacological interventions. For patients with emergency and severe disease, endovascular and surgical interventions can be used to treat patients, aiming for rapid symptom alleviation; however, surgical intervention alone does not significantly improve prognosis. Glucocorticoids (GCs) and methotrexate (MTX) are cornerstone therapies for the management of TAK. More recently, immunotherapy drugs such as tocilizumab have emerged due to a better understanding of the inflammatory mechanisms involved in TAK [1].
The NOD-like receptor family, pyrin domain containing 3 (NLRP3) inflammasome is the most extensively studied inflammasome. It is widely involved in various vascular diseases including atherosclerosis, Kawasaki disease, and anti-neutrophil cytoplasmic antibody-associated vasculitis [8]. Unfortunately, there are limited studies on its relationship with TAK. Recent studies have suggested that the NLRP3 inflammasome and the upstream priming and activation pathways are associated with the severity of TAK [9]. However, the precise underlying mechanisms remain unknown. Herein, this review summarizes the features of TAK as well as the relationship between its pathogenesis and targeting to the NLRP3 inflammasome (Table 1).
| Organ/System | Related diseases |
| Cardiovascular System | CHD, Hypertensive cardiomyopathy, AF, VTE, Ischemic cardiomyopathy, Viral myocarditis, Dilated cardiomyopathy, Diabetic cardiomyopathy, Chemo/radiation-induced myocardial injury, Pericarditis, Vasculitis |
| Respiratory System | COPD, Allergic asthma, COVID-19, Allergic rhinitis, Chronic rhinosinusitis, ALI, Lung cancer, TB, Silicosis, PH, CF |
| Digestive System | Helicobacter pylori infection, Gastritis, NAFLD, Liver cirrhosis, CHB, DILI, Gastric cancer, Liver cancer, IBD, Chronic pancreatitis, Acute pancreatitis, Pancreatic cancer, CHC, AIH, PBC, PSC |
| Nervous System & Psychiatric Diseases | Ischemic stroke, AD, Epilepsy, PD, Hemorrhagic stroke, MS, HD |
| Metabolic System | Obesity, T2DM |
| Kidney Diseases | AKI, DN, Hypertensive nephropathy, LN |
| Connective Tissue & Autoimmune Diseases | RA, Gout, Psoriasis, Atopic dermatitis, CAPS |
| Hematological Diseases | NHL, MM, AML, Lymphoid Leukemia, MDS, GVHD, SCA |
| Infectious Diseases | Sepsis |
| Musculoskeletal System | IVDD |
| Oral Diseases | Periodontitis |
| Ophthalmic Diseases | Allergic conjunctivitis |
| Obstetric Diseases | Preeclampsia |
AD, Alzheimer’s disease; AF, Atrial fibrillation; AIH, Autoimmune hepatitis; AKI, Acute kidney injury; ALI, Acute lung injury; ALL, Acute lymphoblastic leukemia; AML, Acute myeloid leukemia; CAPS, Cryopyrin-associated periodic syndrome; CF, Cystic fibrosis; CHB, Chronic hepatitis B; CHC, Chronic hepatitis C; CHD, Coronary heart disease; CMP, Cardiomyopathy; COPD, Chronic obstructive pulmonary disease; COVID-19, coronavirus disease 2019; DAMPs, Damage-associated molecular patterns; DILI, Drug-induced liver injury; DN, Diabetic nephropathy; GVHD, Graft-versus-host disease; HD, Huntington’s disease; IBD, Inflammatory bowel disease; IVDD, Intervertebral disc degeneration; LN, Lupus nephritis; MDS, Myelodysplastic neoplasms; MM, Multiple myeloma; MS, Multiple sclerosis; NAFLD, Non-alcoholic fatty liver disease; NHL, Non-Hodgkin lymphoma; NLRP3, NOD-like receptor family, pyrin domain containing 3; PAMPs, Pathogen-associated molecular patterns; PBC, Primary biliary cholangitis; PD, Parkinson’s disease; PH, Pulmonary hypertension; PSC, Primary sclerosing cholangitis; RA, Rheumatoid arthritis; SCA, Sickle cell anemia; T2DM, Type 2 diabetes mellitus; TB, Tuberculosis; VTE, Venous thromboembolism.
We conducted a narrative review of the English literature from PubMed, Web of
Science, and Google Scholar databases without strict time restrictions for
eligible publications. The reviewed literature primarily included human clinical
or cellular studies, supplemented with animal experiments investigating relevant
underlying mechanisms. We also incorporated retrospective studies, systematic
reviews, meta-analyses, and narrative reviews. The search utilized the following
keywords: NLRP3 inflammasome, Takayasu arteritis, vasculitis, epidemiology,
incidence, prevalence, burden, pathology, clinical manifestations, pathogenesis,
genetics, genes, inflammation, priming signal, activation signal, Toll-like
receptors (TLRs), interleukins (ILs), IL-1
TAK demonstrates significant epidemiological differences based on ethnicity and region. Previous studies have shown that the incidence and prevalence of TAK are higher in Asian populations, and the majority of patients are young women [5, 10, 11, 12]. A systematic review and meta-analysis including global studies showed a global average incidence of 1.11 (95% CI: 0.70–1.76) cases per million person-years, with a higher prevalence in Asia. The disease is more common in females, with an incidence of 2.01 (95% CI: 1.39–2.90) cases per million person-years (Table 2, Ref. [5, 6, 10, 12]) [13].
| Area | Year | Total population size | Number of cases | Mean annual incidence cases per million (95% CI) | Prevalence cases per million (95% CI) | Age | Male to female ratio | Other findings | Reference number |
| China | 2015 to 2017 | 14,429,700 in 2015, | 102 prevalent cases and 68 incident cases. | 2016 to 2017: 2.33 (1.97–3.21) | 2015 to 2017: 7.01 (5.65–8.37) | Average patient age was 44 years. 16- to 34-year-old subgroup had a prevalence of 11.59 per million and incidence of 3.55 per million. | 1:1.78 | Type V was the most common (38.2%), followed by type I (22.6%), type IIa, (9.8%), type IIb (10.8%), type IV (11.8%), and type III (6.8%). | [5] |
| 14,660,000 in 2016, | |||||||||
| 14,551,300 in 2017. | |||||||||
| Japan | 2017 | Patients with records from 14,291 facilities in Japan. | 2369 | NR | NR | Median age at onset was 29 years. | 1:1.77 | Adult patients had a higher rate (57.8%) of comorbidity than young people (44.9%), with aortic regurgitation being the most common (35.0%). | [6] |
| The affected rate of each artery: common carotid artery and internal carotid artery (62.7%), subclavian artery (57.8%), arch of aorta, (49.8%), and thoracic descending aorta (40.0%). | |||||||||
| US | 2010 to 2018 | Patients with records resided in 27 counties in the US. | 5 prevalent cases and 1 incident case. | NR | 2010 to 2015: 8.4 (1–15.8) | Mean age at diagnosis was 20.5 years. | 1:3.61 | Type V was the most common (80.0%), followed by type I (20.0%). | [10] |
| UK | 2000 to 2005 | About 3.6 million patients and 445,000 patients from 2 different databases. | 16 prevalent cases and 14 incident cases from the UKGPRD. | 0.8 (0.4–1.3) | 7.1 (NR) | Median age at diagnosis was 51.0 years. | 1:13.00 | The incidence of TAK in the UK is similar to that in other countries. | [12] |
CI, Confidence interval; NR, Not reported; TAK, Takayasu’s arteritis; UKGPRD, UK General Practice Research Database.
TAK can be divided into two main phases: active (systemic/acute) phase and inactive phase. In the initial stage of the active phase, inflammation primarily affects the vasa vasorum and the junction between the media and adventitia. Mononuclear cell infiltration, cellular edema, elastic fiber rupture, granuloma formation, and necrosis of the media can be observed. In the developing stage, there is reactive fibroplasia in the intima, and neovascularization occurs between the intima and media. Severe inflammatory reactions can induce the death of smooth muscle cells in the media, leading to gradual dilation of the vessels and even the formation of aneurysms. After entering the inactive phase, fibrosis and scar formation occur in the adventitia, and the infiltrating inflammatory cells mainly consist of plasma cells and multinucleated giant cells. Fibroplasia and granuloma formation compress the lumen, resulting in stenosis and the appearance of symptoms [14].
TAK can have an insidious onset or present acutely, which manifests as acute ischemic necrosis such as stroke or myocardial infarction [15]. Symptoms depend on which blood vessels are affected. When the renal arteries are involved, hypertension can occur. Severe narrowing or occlusion of the blood vessels in a short period of time can lead to a hypertensive crisis. This sudden and significant increase in blood pressure can cause various symptoms such as headache and nausea [16, 17]. Aortic valve regurgitation is a significant complication of TAK and can lead to heart failure, which is a main cause of death in patients with TAK [18]. TAK involvement of the coronary arteries is rare, with an incidence of 6–30% in patients and a 3% chance of causing acute myocardial infarction [19, 20]. TAK is a cause of renal impairment in children and can also contribute to growth retardation [21, 22]. Increased erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) are commonly observed in patients with TAK. However, it is noteworthy that only less than 20% of patients show an increased white blood cell count (Fig. 1, Ref. [23, 24, 25, 26, 27, 28]) [29].
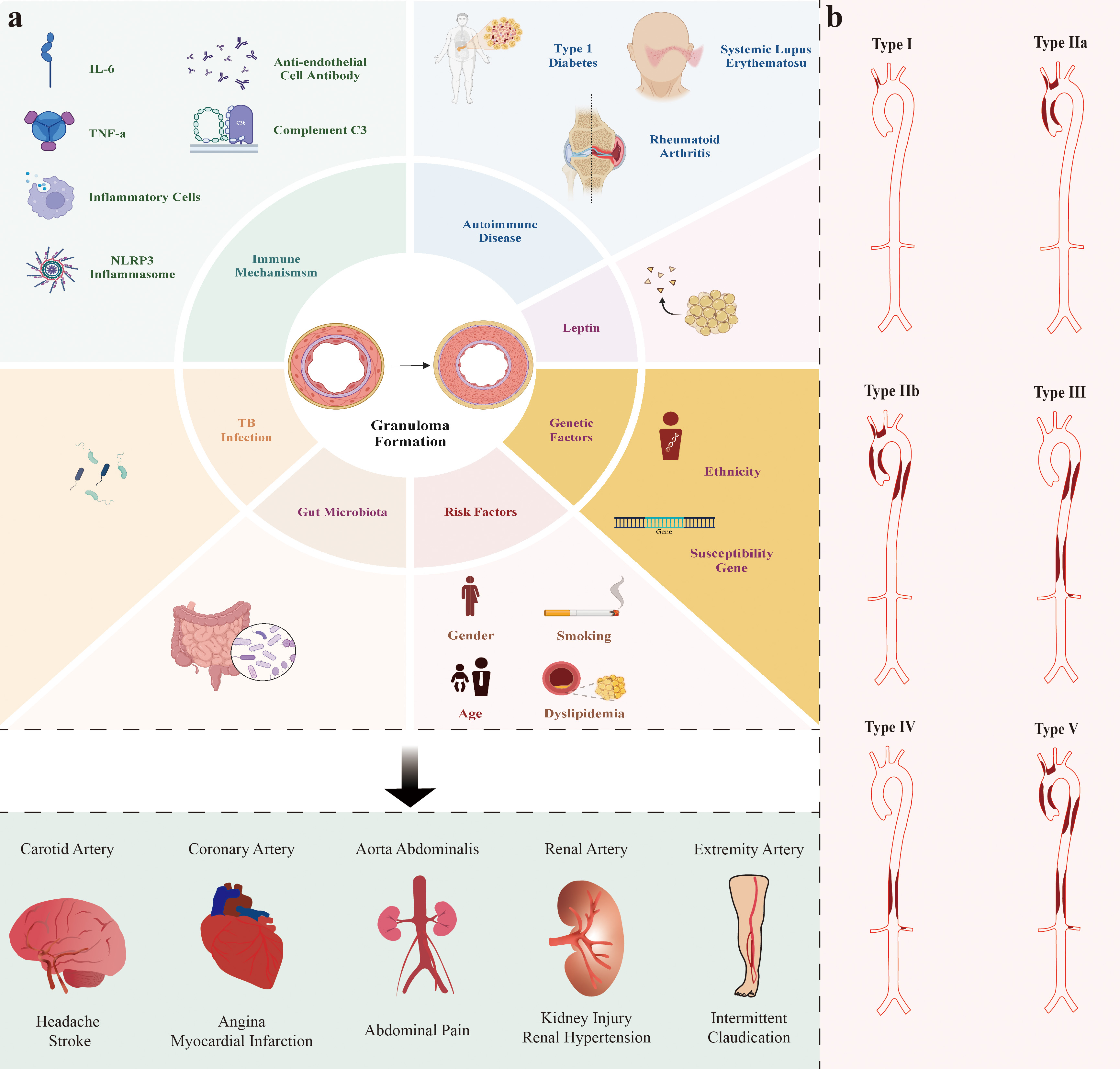 Fig. 1.
Fig. 1.
Risk factors and potential pathogenesis/mechanisms involved in TAK and Hata’s angiografic classification of TAK. (a) The risk factors and elements involved in the pathogenesis of TAK are not yet fully understood. Studies generally suggest that risk factors for TAK include sex (possibly related to estrogen levels), age, dyslipidemia, and smoking [23, 24, 25]. The pathogenesis of TAK involves multiple aspects such as genetics, immunity, and M. tuberculosis infection. Recent studies indicate that the pathogenesis of TAK may also be associated with new factors such as the gut microbiome and leptin level [26, 27]. It is also thought that TAK itself is an autoimmune disease, indicating a common underlying mechanism. In summary, different arterial involvements cause corresponding symptoms. (b) Hata’s classification is widely used in clinical practice to categorize TAK based on vessel involvement, which includes aortic arch, ascending aorta, thoracic descending aorta, abdominal aorta, renal arteries and their branches [28].
For stenosis, vascular intervention treatment is considered the routine option; however, patients with TAK are prone to restenosis after this intervention due to the persistent inflammatory response inherent in this disease. The in-stent restenosis rate is as high as 60% in patients with TAK, but only 5–10% in patients with coronary heart disease [30, 31]. Clinicians should be aware of the aforementioned complications associated with TAK and the importance of combined anti-inflammatory treatment, as routine interventional therapy often yields poor results.
The current pathogenesis theory suggests that the persistent inflammatory
response leading to TAK may be caused by microbial antigens [32]. Increasing
studies point to a new understanding that the vascular system may not be
completely sterile, and immune reactions to pathogenic protein antigens may
cross-react with natural proteins on host vascular tissue cells [33]. Kumar Chauhan S
et al. [34] found elevated levels of HSP65 in the arterial tissue of patients
with TAK. HSP65 is found in pathogens such as M. tuberculosis and
is highly similar to HSP60, which is widely present in human tissue cells.
Patients with TAK develop specific antibodies recognizing both HSP60 and HSP65
[26]. Antigens produced by pathogens, such as lipopolysaccharide (LPS), may be
recognized by antigen-presenting cells (APCs) and the pathogen’s exotoxins can
upregulate the expression of TLRs on APCs and mononuclear-macrophages. Activation
of T cells, B cells, and natural killer (NK) cells induces the apoptosis of cells
through various pathways. Additionally, HSP65 stimulates arterial tissue cells to
synthesize MHC class I polypeptide-related sequence A proteins, which bind to
NKG2D receptors on
Studies suggest that TAK has genetic factors influencing susceptibility and is prevalent in individuals of Asian and Slavic descent, including those who have immigrated to Western countries, compared to the local white population [12, 38]. The HLA-B52 gene is the most well-known related genetic factor, and is more prevalent in high-incidence Asian populations compared to European populations. Meanwhile other newly discovered risk factors include sushi, von Willebrand factor type A, epidermal growth factor (EGF) and pentraxin domain containing 1, cofilin 2, VPS8, chr13q21, and the IL18-137G/C polymorphism, while protective factors include the IL18-607C/A polymorphism and killer cell immunoglobulin-like receptor 2DS4 gene; however, further studies are needed to determine the ethnic distribution of these genes (Fig. 1) [39, 40, 41, 42, 43, 44, 45, 46].
The activation of inflammasomes is closely related to many diseases with an
inflammatory component. The inflammasome causes cell pyroptosis and participates
in the innate immune response, triggering local as well as systemic inflammatory
responses. Activation of the inflammasome occurs by the binding of cysteinyl
aspartate specific proteinase 1 precursor (pro-caspase 1) to pattern recognition
receptors (PRRs) through apoptosis-associated spot-like protein (ASC). PRRs,
particularly the NOD-like receptor family (e.g., NLR family pyrin domain
containing 1 [NLRP1], NLRP2, NLRP3, NLRP6, NLRP12, NLR family caspase recruitment
domain (CARD) domain-containing protein 4 [NLRC4]), are key players in the immune
system’s ability to detect and respond to threats. PRR can initiate pathways that
lead to the activation of pro-caspase-1 and its subsequent regulation of
IL-1
The canonical pathway for NLRP3 inflammasome activation generally requires two
distinct signals: a priming signal and an activation signal. The priming signal
(signal I), involves the upregulation of NLRP3 and pro-IL-1
There is evidence supporting the idea that HSP65 is highly expressed in vascular
tissues and may act as a priming signal as a TLR agonist [51]. It is a member of
the HSP60 family, mainly presenting in M. tuberculosis and originally
isolated from My. bovis. HSP65 shares a high degree of similarity with
human HSP60 [52]. Additionally, HSP60 can activate macrophages by interacting
with TLRs, initiating the synthesis of NLRP3 and triggering inflammatory cascades
[53]. Huang et al. [54] found that in patients with gouty arthritis,
HSP60 expression was increased in peripheral blood mononuclear cells (PBMCs) and
HSP60 activated the TLR4/myeloid differentiation primary response protein 88
(MyD88)/NF-
TNF-
According to current understanding, activation signals may involve multiple
factors. After cell perforation, DNA fragments, specifically extracellular dsDNA
(eDNA), are released from cells undergoing death due to the activity of
Also, an alternative activation pathway of NLRP3 inflammasomes exists in human
monocytes. The binding between LPS and TLR4 can stimulate the assembly of NLRP3
inflammasomes, and this can occur through a pathway involving
receptor-interacting serine/threonine-protein kinase 1, Fas-mediated death domain
protein, and caspase-8, bypassing the priming step [61]. It has also been found
that TLR4 recognizes LPS in the PBMCs of patients with TAK, which increases the
expression of IL-1
In 2014, Maejima et al. [9] found a correlation between the severity of symptoms in patients with TAK and polymorphism of the MAX Dimerization Protein MLX (MLX) gene. Specifically, patients with TAK with severe symptoms had a higher frequency of the missense mutation rs665268 in the MLX gene, indicating a potential association between MLX protein and TAK pathogenesis [9]. Previous studies have established a correlation among MLX protein, TXNIP, and NLRP3 inflammasome activation, particularly under conditions of oxidative stress and involving the mitochondria [64, 65]. Research suggests a link between MLX allelic variants and the NLRP3 inflammasome, which plays a role in the pathogenesis of TAK. In 2014, a study by Maejima et al. [9] confirmed that the MLX risk genotype upregulates MLX protein transcription, which in turn promotes the expression of TXNIP through the MLX/Mondo-A complex. The authors also observed an increase in NLRP3 inflammasome and caspase-1 levels in the cultured macrophages of patients with TAK harboring this allele compared to the control group [9].
Additionally, this genotype was associated with decreased autophagy levels, possibly due to reduced free Mondo-A resulting from its binding to the MLX protein. Mondo A promotes autophagy by inhibiting the expression of Rubicon, which in turn acts as a brake on autophagy, particularly by inhibiting the formation of auto-phagolysosomes (autophagosomes fused with lysosomes for degradation). Rubicon accomplishes this by binding to the phosphoinositide 3-kinase complex and inhibiting its activity [66].
Mitophagy plays a crucial role in the negative regulation of the NLRP3 inflammasome by clearing damaged mitochondria, one of the sites that generate upstream activation signals for NLRP3. Damaged mitochondria release ROS and oxidized mitochondrial DNA into the cytosol, which induce activation of the NLRP3 inflammasome through mechanisms that are not fully understood [67]. Moreover, the NLRP3 inflammasome can be engulfed by autophagosomes through ubiquitination and subsequent degradation. Ubiquitination can activate autophagy, resulting in the inactivation of NLRP3 inflammasomes [68].
Macro-autophagy and ROS act as intermediaries in the correlation between STAT3
and NLRP3. Phosphorylated STAT3 acts as a transcriptional activator, binding to
target genes and promoting their expression. In mice, activation of STAT3
increases the acetylation of histones H3 and H4 on the NLRP3 promoter, thereby
promoting the expression of effector proteins. Applying JAK inhibitors to reduce
STAT3 phosphorylation will continuously inhibit NLRP3 transcription, suppressing
inflammasome expression [69]. In addition, stimulating the marrow-derived
macrophages of mice with LPS showed that STAT3 inactivation blocked NLRP3
inflammasome-induced caspase-1 activation and IL-1
One of the activation pathways of NLRP3 inflammasome is mediated by the P2X7R
starting with signal II [47, 71]. P2X7R is a transmembrane receptor, and ATP is
its only physiological agonist [72]. It is widely expressed in various cells of
the human body including endothelial cells, fibroblasts, neutrophils,
monocytes/macrophages, dendritic cells, and lymphocytes [73]. Binding of the
P2X7R with its agonist causes potassium efflux, sodium and calcium influx, and
the formation of non-selective pores that allow the passage of macromolecules
[74]. Cell damage or death leads to the release of ATP, which activates the
P2X7R, resulting in the variation of these electrolytes and the entry of PAMPs
through the pores [75, 76]. These actions disrupt the cellular homeostasis,
leading to organelle damage, ROS release, initiation of the ROS-NLRP3 axis, and
assembly of the NLRP3 inflammasome [77]. P2X7R can also mediate the release of
IL-1
Monocytes/macrophages with high expression of P2X7R are found in the vascular wall [80]. P2X7 polymorphisms are associated with both TAK and tuberculosis infection. The P2X7-1513C is a mutation that leads to non-functional P2X7R expression in macrophages. The frequency of this polymorphism is higher in patients with TAK than those with only pulmonary tuberculosis, and tuberculosis genetic material can also be detected in the aortic arch and other arterial tissues of patients with TAK [81]. Theoretically, the extent of pyroptosis mediated by the eATP-P2X7R pathway in the macrophages of patients with TAK would be weakened according to this finding, which would partially compromise the innate immunity against M. tuberculosis. It has been hypothesized that the proliferation of M. tuberculosis through the aforementioned mechanism can lead to the increased synthesis of HSP65 protein and exacerbate TAK.
Cells release two main inflammatory mediators, IL-1 and IL-18, after NLRP3
inflammasome activation. IL-1 has strong inflammatory-inducing effects that can
lead to prostaglandin secretion and activate and differentiate neutrophils,
monocytes, and lymphocytes [82]. IL-18 is a key player in inducing IFN-
As aforementioned, there is a close relationship between IL-6 and TAK. The
levels of IL-6 in the peripheral blood and lesion tissues of patients with TAK
are significantly elevated compared to healthy individuals, and IL-6 is
significantly involved in the pathogenesis of TAK. First, IL-6 is closely
associated with changes in inflammatory markers such as ESR and CRP, which can
reflect the severity of inflammation during the acute phase of the disease.
Second, in the early stage of TAK, monocytes and fibroblasts are activated
through the TLR/NF-
Researchers have observed in a mouse model of rheumatoid arthritis (RA) that
IL-6 can activate the NLRP3 inflammasome in the presence of ATP. Conversely,
blocking the IL-6 pathway leads to reduced NLRP3 inflammasome [62]. The two major
downstream signaling pathways activated by IL-6/IL-6R are JAK/STAT3 and
JAK/mitogen-activated protein kinase. Studies have shown that IL-6 may also
influence the initiation of NLRP3 inflammasome mediated by NF-
Research on lung tissues infected with SAR-CoV-2 has revealed a cascade reaction
of NLRP3/IL-1
The inflammation response in TAK involves a complex interplay between neutrophil extracellular traps (NETs) and NLRP3 inflammasome activation. NETosis is a process in which neutrophils, upon external stimulation, undergo disintegration of nuclear membranes and decondensation of chromatin structures to form fibrous extracellular networks called NETs. These NETs are composed of DNA, citrullinated histone H3 (CitH3), and other proteins, which are subsequently released into the extracellular space. NETs play a dual role in the immune defense by trapping, containing, and even killing pathogens in the extracellular environment, while simultaneously exacerbating inflammatory responses [93].
Suicidal NETosis, one form of NETosis, is triggered by stimuli such as IL-6,
IL-8, TNF-
In patients with TAK, researchers have observed that NET levels correlate with
disease activity, and serum samples exhibit the presence of anti-citrullinated
protein/peptide antibodies (ACPA) targeting CitH3 [96]. In other diseases or
animal models, factors implicated in TAK pathogenesis such as IL-6, TNF-
Activation of the NLRP3 inflammasome facilitates the production of NETs. The
formation of inflammasome-associated ASC is observed prior to chromatin
decondensation, and the presence of ASC and caspase-1 associated with CitH3 is
also noted in NETs formed in other specific diseases [101]. In the disease model
of gout, the absence of Caspase-11, downstream of the non-canonical NLRP3
pathway, restricts the generation of neutrophil NETs, whereas which should formed
under the stimulation of monosodium urate (MSU) [102]. IL-1
In summary, TAK involves both NETosis and NLRP3 inflammasome activation, with NLRP3-mediated regulation to NETosis observed in other diseases. However, no studies to date have directly determined whether neutrophil-derived NETs and NLRP3 inflammasome activation co-occur in TAK. Analyzing their interplay faces challenges due to the disease’s complex and unclear mechanisms, making it difficult to determine the temporal sequence between NETosis and NLRP3 activation. Moreover, numerous mechanistic questions remain unresolved, such as whether NET components like CitH3 and dsDNA trigger NLRP3 assembly or whether GSDMD-mediated pyroptosis amplifies NETosis in TAK. Further research is needed to elucidate these interactions and their contribution to vascular inflammation.
The pharmacotherapy used to treat TAK includes using GCs, immunosuppressive
agents, and newer targeted biologic drugs like tocilizumab and infliximab [1, 104]. GCs are well-known anti-inflammatory agents and are recommended by the
American College of Rheumatology (ACR) as first-line therapy for TAK [1]. Using
GCs alone can achieve clinical remission for 60% of patients, but relapse is
common after dosage reduction [105]. Studies have found that GCs can inhibit the
activity of NLRP3 inflammasomes, reducing levels of IL-1
Although GC therapy is a cornerstone treatment for some patients, some individuals either do not achieve full remission or experience relapses. In such cases, targeted drugs offer several advantages such as reduced hormone dosage, enhanced remission rates, and decreased recurrence. Major targets for targeted drug therapies in TAK include blocking TNF receptors, TLR4, IL-6Rs, and the JAK/STAT3 pathway (Fig. 2).
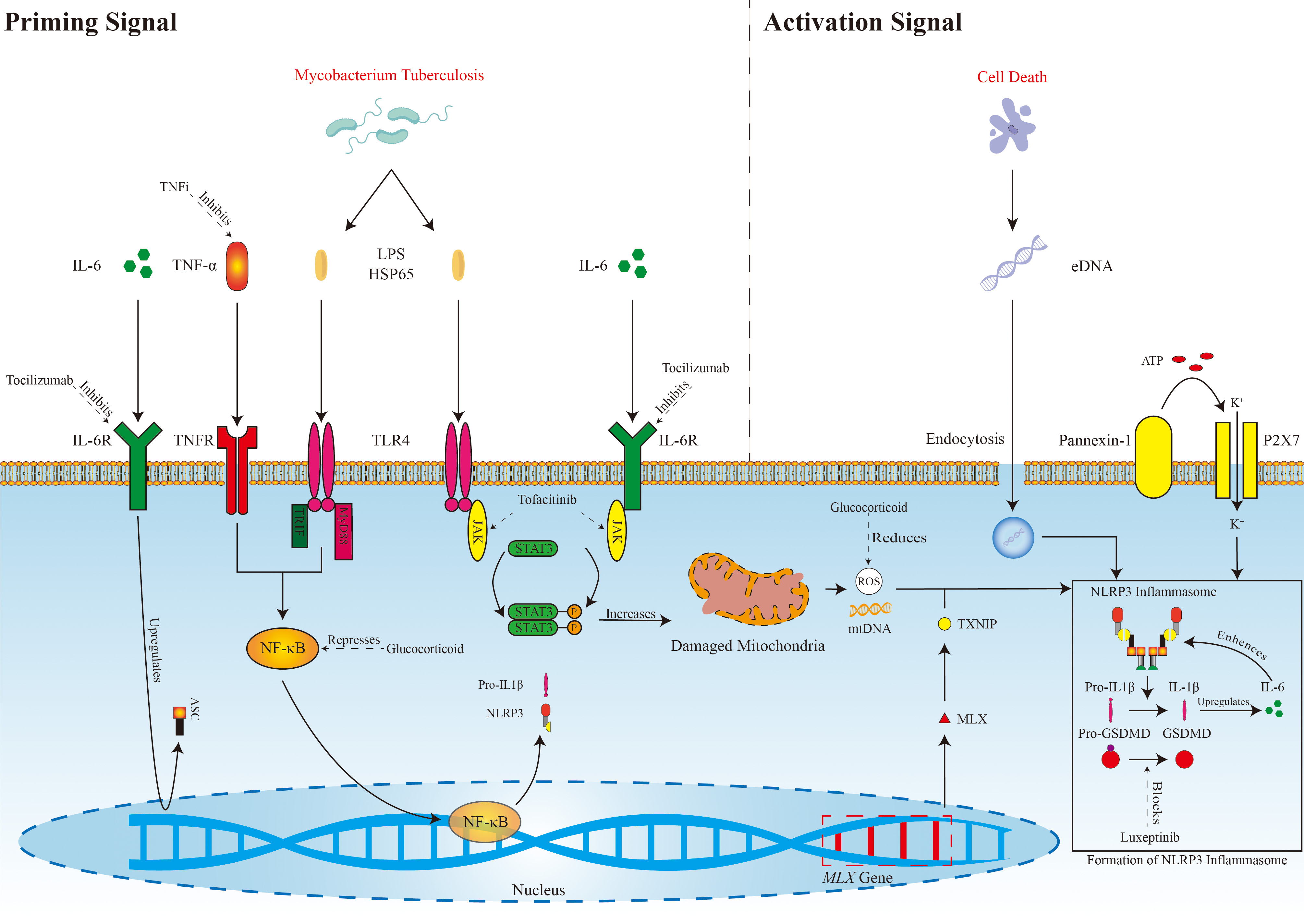 Fig. 2.
Fig. 2.
Possible mechanisms of NLRP3 inflammasome activation in TAK and
targeting pathways.
TNFi, Tumor necrosis factor alpha inhibitor; IL-6, Interleukin-6;
TNF-
Infliximab is a TNF-
The formation of NLRP3 inflammasomes in TAK involves the canonical pathway
mediated by a priming signal and activation signal. TNF-
Etanercept is another type of TNF inhibitor used for the treatment of TAK; it
blocks the effects of TNF-
Tocilizumab is an IL-6R monoclonal antibody, and the NLRP3 inflammasome may be a
crucial component in TAK. Targeting this pathway with drugs like tocilizumab may
be a relevant treatment strategy that could affect ASC synthesis, the clearance
of damaged mitochondria, or even directly inhibit the function of NLRP3 by
blocking the IL-6R, as discussed previously. Tocilizumab not only blocks the
IL-6R but also inhibits activation of the NLRP3 inflammasome through the IL-17a
pathway, leading to a significant decrease in IL-17a expression [111]. IL-6
promotes the differentiation and formation of Th17 cells, and the secretion of
cytokines such as IL-17 and TNF-
Ziltivekimab is a novel IL-6 ligand human monoclonal antibody that alleviates inflammation and reduces serum CRP levels. It was developed to provide a new option for the anti-inflammatory treatment of atherosclerosis in patients with chronic kidney disease, as increasing data suggest that the pro-inflammatory effects of IL-6 can exacerbate atherosclerosis and thrombosis. Studies have found that regular use of this drug, with subcutaneous injections of 7.5, 15, or 30 mg every 4 weeks for 24 weeks, can effectively lower CRP levels in patients with significant inflammatory responses [117]. It is currently unknown whether ziltivekimab has a positive effect on cardiovascular outcomes, as clinical trial results are not yet available. To date, ziltivekimab has not been used for the treatment of TAK and its therapeutic efficacy remains to be substantiated.
TAK-242 (also known as Resatorvid) is a specific inhibitor of TLR4 signaling that shows promise for alleviating diseases such as ulcerative colitis [118, 119]. Wu et al. [58] used TAK-242 to block HSP65 activation of the TLR4-JAK2/AKT/STAT3 pathway and reduce the phosphorylation levels of JAK2, AKT, and STAT3 in aortic adventitial fibroblasts in vitro, showing a possibility as a targeted therapy. However, for treating other indications like sepsis, TAK-242 is either undergoing Phase 3 clinical trials or the trial results are unsatisfactory, limiting its clinical application.
JAK inhibitors work by blocking the JAK/STAT3 signaling pathway. Traditionally, it is believed that JAK inhibitors achieve therapeutic effects by reducing Th1- and Th17-derived cytokines and activating CD4+ cells. Therefore, JAK inhibitors may also influence the initiation and activation signals of the NLRP3 inflammasome through two mechanisms: blocking STAT3 upregulation of NLRP3 protein expression and enhancing the clearance of damaged mitochondria. Clinical trials have shown that the combined treatment with drugs such as tofacitinib and ruxolitinib for the treatment of TAK yields better results than using GCs alone [120, 121]. Currently, several clinical trials are underway (e.g., NCT04161898 and NCT04299971) (Table 3, Ref. [58, 109, 115, 120, 121, 122, 123, 124, 125, 126, 127]).
| Reference | Target factor | Study design | Result | Reference number |
| Mertz et al. | TNF- |
Clinical trial | Infliximab could be an effective GC-sparing agent for TAK refractory to conventional therapy. | [122] |
| 2020 | IFX | 23 patients from 12 centers | Rate of response after a median treatment duration of 36.9 months (IQR: 10.0–58.7): 64% | |
| Median dose of GCs at initiation: 10 mg/day (IQR: 10–25). | GC dose decreased from median 10 mg/day (range 5–45 mg/day) at baseline to less than 10 mg/day at 12 months (p |
|||
| Median dose of infliximab: 5 mg/kg (IQR: 5–5) every 6 weeks (IQR: 4–9). | ||||
| Mekinian et al. | TNF- |
Retrospective study | Infliximab may represent an interesting alternative therapeutic option even in refractory TAK. | [123] |
| 2012 | IFX | 15 patients | Rate of response at 3 months: 87% | |
| Median dose of GCs at initiation: 20 mg/day (range 5–35 mg/day). | Rate of response at 6 months: 77% | |||
| Rate of response at 12 months: 73% | ||||
| Median dose of infliximab: 5 mg/kg (range 3–5 mg/kg) at a median of every 6 weeks (range 4–8 weeks). | Clinical and biological activities significantly decreased within 3 months (from 11 at baseline to 4 patients at 12 months; p |
|||
| GC dose decreased from median 20 mg/day (range 5–35 mg/day) at baseline to median 6 mg/day (range 2.5–30 mg/day) at 12 months (p |
||||
| Misra et al. | IL-6 receptor and TNF- |
Systematic review and meta-analysis | Tocilizumab and TNFi had similar rates of clinical remission (RR [tocilizumab vs. TNFi]: 1.03, 95% CI 0.91–1.17), angiographic stabilization (RR 1.00, 95% CI 0.72–1.40) or adverse events (RR 0.84, 95% CI 0.54–1.31) based on observational data. | [124] |
| 2023 | TCZ and TNFi | 6 studies and 491 patients were included. | ||
| Kang et al. | IL-6 receptor | Systematic review and meta-analysis | Rate of decrease in GC dosage was 76% (95% CI 58–87%). Remission rate was 79% (95% CI 69–86%). Relapse rate was 17% (95% CI 5–45%). Imaging progress rate was 16% (95% CI 9–27%). Adverse events occurred in 16% (95% CI 5–39%) of patients, and infection was the most common adverse event, with a rate of 12% (95% CI 5–28%). | [125] |
| 2023 | TCZ | 19 studies and 466 patients were included. | ||
| Wang et al. | IL-6 receptor and TNF- |
RCT | ADA combined with GCs and MTX may be more efficacious than TCZ combined with GCs and MTX in patients with active and severe TAK. | [109] |
| 2024 | TCZ and ADA | 40 patients were included. | ||
| ADA (n = 21) combined with GCs and MTX vs. TCZ (n = 19) combined with GCs and MTX. | ERs of the ADA group and TCZ group at 6 months were 85.71% (p = 0.02) and 52.63% (p = 0.02) in the ITT population, 89.47% (p = 0.06) and 62.50% (p = 0.06) in the PPS. The ERs at 9 and 12 months were similar (p |
|||
| Nakaoka et al. | IL-6 receptor | RCT | TCZ was superior to placebo for time to relapse of TAK and steroid-sparing effect without new safety concerns. | [115, 126] |
| 2018 and 2020 | TCZ | 36 patients (18 received TCZ s.c. 162 mg/week and 18 received placebo after remission from TAK for |
||
| In the double-blind period, HRs for time to relapse were 0.41 (95.41% CI 0.15–1.10; p = 0.0596) in the ITT population (primary endpoint) and 0.34 (95.41% CI 0.11–1.00; p = 0.0345) in the PPS | ||||
| In the open-label extension period, 46.4% of patients reduced the dose, which was less than one-half the dose administered at relapse before study entry. | ||||
| Wang et al. | JAK/STAT3 | RCT | LEF and TOF were comparable for TAK treatment. TOF group had advantage of GC dose reduction while keeping a similar rate of persistent remission (46.88% vs. 17.14%) | [120] |
| 2022 | TOF | GC in addition to TOF vs. GC in addition to LEF | ||
| Kong et al. | JAK/STAT3 | RCT | TOF group had a greater advantage in CR rate (88.46% vs. 56.52%; p = 0.02), relapse rate (11.54% vs. 34.78%; p = 0.052), median relapse-free duration (11.65 |
[121] |
| 2022 | TOF | GC in addition to TOF vs. GC in addition to MTX | ||
| No statistical difference in side effects between the two groups. | ||||
| Wu et al. | TLR4-JAK2/AKT/STAT3 | Clinical trial | Patients with Kerr score |
[58] |
| 2021 | Curcumin | Curcumin (7.5 g bid) in addition to GC, tacrolimus, LEF, AZA, MMF, Rapamycin and HCQ | ||
| Régnier et al. | JAK/STAT3 | Clinical trial | CD4+ effector T-cell activation or differentiation was reduced, expression by CD4+ T cells was reduced and Treg percentages were increased after 6 months of treatment compared with baseline. | [127] |
| 2020 | JAKi | GC and JAKi | ||
| CRP level at 6 months was reduced and treatment allowed GC dose reduction for two-thirds of patients and led to a reduction of NIH activity score to 0 for all treated patients compared with baseline. |
ADA, Adalimumab; AZA, Azathioprine; CI, Confidence interval; CR, Complete remission; CRP, C-reactive protein; ER, Efficacy rate; GC, Glucocorticoid; HCQ, Hydroxychloroquine; HR, Hazard ratio; IFX, Infliximab; IQR, Interquartile range; ITT, Intention-to-treat; JAKi, JAK inhibitor; LEF, Leflunomide; MMF, Mycophenolate mofetil; MTX, Methotrexate; NIH, National Institutes of Health; PPS, Per-protocol set; RCT, Randomized controlled trial; RR, Risk ratio; s.c., Subcutaneous; TCZ, Tocilizumab; TAK, Takayasu’s arteritis; TNFi, Tumor necrosis factor alpha inhibitor; TOF, Tofacitinib.
IL-1
In conclusion, TAK, is a rare but serious and complex inflammatory disease with a still largely unknown pathogenesis, posing a significant challenge to human health. While progress has been made in understanding the risk factors, pathogenesis mechanisms, pathological changes, and clinical features in TAK in recent years, the specific role of the NLRP3 inflammasome in the development of TAK needs further in-depth investigation to improve our knowledge and promote a novel targeted therapeutic strategy.
Recent studies have revealed the significant role of NLRP3 inflammasomes in various rheumatic diseases such as gouty arthritis. In comparison, TAK is rarer but has an equally profound impact, particularly on the younger population, with potential risks of disability and sudden death. In 2018, it was first confirmed that NLRP3 inflammasomes are expressed at elevated levels in cultured macrophages from patients with TAK. However, a limited number of follow-up studies and theoretical updates have been pursued since that time and numerous questions remain unanswered. First, HSP60 has been established as a priming signal for the NLRP3 inflammasome, and there is evidence suggesting that HSP65, due to its homology with HSP60, might also be involved in regulating the priming of NLRP3 inflammasomes, possibly through TLR4-related pathways. In addition, in vitro experiments have demonstrated that the MLX genotype diversity indirectly modulates the activation of NLRP3 inflammasomes by affecting oxidative stress mechanisms; however, there is a lack of animal model validation of this finding. MLX genotype does influence the process, but it remains unknown if MLX gene diversity plays a predominant role in the current prevalence of TAK. Furthermore, an interesting observation is that functional P2X7Rs can mediate the activation of NLRP3 inflammasomes, whereas non-functional receptors exhibit a higher expression frequency in patients with TAK combined with pulmonary tuberculosis. The P2X7R is involved in both the immune response to eliminating M. tuberculosis and plays a role in mediating the activation of NLRP3 inflammasomes. It remains unknown how the P2X7R and NLRP3 inflammasomes influence the occurrence of TAK combined with pulmonary tuberculosis. Existing theories still contain gaps that require in-depth investigations (Table 4). The study of these mechanisms will enhance the understanding of TAK and inflammation, potentially accelerating the discovery of novel therapeutic targets and interventions.
| Current status | Unmet needs | Proposals and potential solution |
| Limited epidemiological data due to low incidence and prevalence rates of TAK | Analysis of the temporality between risk factors and disease onset | Implementation of a cohort study |
| High underdiagnosis rate of TAK due to nonspecific clinical presentation | Enhancement of clinician awareness of TAK | Screening of patients with similar clinical manifestations using angiography |
| Complexity of current induction methods and suboptimal fidelity in animal models | Animal models demonstrating superior hit rate, cost-effectiveness, and survival rate | Exploration and refinement of animal model establishment |
| Under investigated hereditary patterns of newly identified susceptibility genes | Lack of inheritance patterns and population distribution of emerging genetic variants including SVEP1, CFL2, VPS8, chr13q21, and IL18-137G/C | Pedigree investigation and genetic sequencing of probands |
| Unclear role of HSP65 in linking HSP60, M. tuberculosis, and TAK pathogenesis | Investigation of M. tuberculosis pathogenesis via the connection with HSP65 and HSP60 directly in TAK | Examining TLR4-mediated signaling pathway activation by stimulating in vitro-cultured neutrophils from TAK patients with HSP65 |
| Limited in vitro evidence of the MLX gene in modulating NLRP3 inflammasome formation via oxidative stress in TAK | Delineation of the ROS-TXNIP-NLRP3 axis pathogenic role within TAK | Assessment of MLX-knockout animal model manifestations of the pathological features of TAK |
| Non-functional P2X7R may reduce NLRP3 inflammasome activation signals and confer immune privilege to M. tuberculosis, exhibiting a paradoxical dual relationship in TAK pathogenesis; however, direct experimental evidence remains lacking. | Research on the effect of non-functional P2X7R on NLRP3 inflammasome activation in a TAK model | Measure NLRP3 levels in neutrophils derived from patients with TAK following P2X7R blockade. |
| Research on the relationship between non-functional P2X7R affecting M. tuberculosis clearance and TAK-related manifestation | Assess the pathogen clearance of TAK patient-derived P2X7R blocked macrophages after infecting with M. tuberculosis. |
HSP65, Heat shock protein 65; P2X7R, P2X7 receptor; ROS, Reactive oxygen species; TAK, Takayasu’s arteritis; TLR4, Toll-like receptor 4; TXNIP, Thioredoxin-interacting protein.
ACPA, anti-citrullinated protein/peptide antibodies; APLs, altered peptide ligands; APCs, antigen-presenting cells; ASC, apoptosis-associated spot-like protein; CitH3, citrullinated histone H3; CRP, c-reactive protein; dsDNA, double-stranded DNA; ESR, erythrocyte sedimentation rate; eATP, extracellular ATP; eDNA, extracellular dsDNA; GSDMD, Gasdermin D; GC, glucocorticoid; HSP65, heat-shock protein 65; IL, interleukin; MMPs, matrix metalloproteinases; MTX, methotrexate; MPO, myeloperoxidase; NK cells, natural killer cells; NE, neutrophil elastase; NETs, neutrophil extracellular traps; NLRP3, NOD-like receptor pyrin domain-containing protein 3; PAMPs, pathogen-associated molecular patterns; PRR, pattern recognition receptor; PBMCs, peripheral blood mononuclear cells; PINK1, PTEN-induced kinase 1; P2X7R, P2X7 receptor; ROS, reactive oxygen species; RIPK1, receptor-interacting serine/threonine-protein kinase 1; RA, rheumatoid arthritis; TAK, Takayasu’s arteritis; TXNIP, thioredoxin-interacting protein; TLRs, Toll-like receptors; TNF, tumor necrosis factor.
ZYW, TYM, KH and JHZ conceived and contributed to the writing of the main manuscript text. ZYW, TYM and KH prepared figures and tables. SJL and JJL designed the review scope and objectives, supervised the interpretation of critical findings, and also reviewed and revised the paper. All authors reviewed the manuscript and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
The authors are supported by the specific research fund of the Innovation Platform for Academicians of Hainan Province: Dynamic monitoring and genetic correlation of blood biochemical indexes related to cardiovascular and cerebrovascular diseases in “migratory bird population” in Hainan (No. YSPTZX202032), Hainan Province Science and Technology Special Fund (ZDYF2020213) and Natural Science Foundation of Hainan (821QN425).
The authors declare no conflict of interest. Jian-Jun Li is serving as one of the Editorial Board members of this journal. We declare that Jian-Jun Li had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Allison B. Reiss.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.