
- Academic Editor
Inherited cardiac arrhythmias, which may lead to sudden cardiac death, represent a significant health risk, with genetic factors playing a key role in their development. The ankyrin 2 (ANK2) gene, encoding ankyrin-B, is implicated in several heritable arrhythmia syndromes. ANK2 variants have been linked to an inherited condition known as “ankyrin-B syndrome”, which manifests as a spectrum of cardiac arrhythmias and cardiomyopathy. Our current review examines the relationship between ANK2 variants and specific heart conditions, summarizing recent findings on the genetic and molecular mechanisms underlying ANK2-related arrhythmias and structural abnormalities. By emphasizing the need for further research, this review aims to enhance understanding of ANK2’s role in heart disease and guide the development of effective therapies.
Cardiac arrhythmias are common and potentially lead to sudden cardiac death (SCD) [1]. A previous study has linked genetic variants in ion channel genes to an increased risk of inheritable arrhythmia syndromes [2]. These genes are crucial for regulating the flow of electrically charged ions in and out of cells. Variants in these genes can affect the biophysical properties and trafficking of ion transporters and channels. A recent study has expanded the understanding of arrhythmias by focusing on genetic variants in non-ion channel genes [3]. One such gene is ankyrin 2 (ANK2), which is responsible for coding a protein known as ankyrin-B [4, 5, 6, 7].
Ankyrin-B, a member of the ankyrin protein family [7], is a protein that serves as an adapter essential for the proper expression and targeting of various integral membrane proteins in the heart, including cardiac ion channels, transporters, receptors, and signaling molecules [5]. The canonical structure of 220-kD ankyrin-B consists of several functional domains [8]. The N-terminal membrane-binding domain (MBD) comprises 24 ANK repeats, which are highly conserved and are involved in interactions between proteins. The spectrin-binding domain (SBD) is composed of a tandem of Zinc-binding domain 5 N-terminal-Zinc-binding domain 5 C-terminal-UPA (ZU5N-ZU5C-UPA) domains, facilitating interactions with a cytoskeletal protein called spectrin. The regulatory domain (RD) includes a death domain (DD) and a variable C-terminal domain (CTD) [8].
In humans, variants in the ANK2 gene that result in the loss of function of ankyrin-B protein can significantly impact the activity of ion channels, leading to electrical instability in the heart. These variants have been linked to an inherited condition known as “ankyrin-B syndrome (ABS) [5, 9]”, which manifests as a spectrum of cardiac arrhythmias and can lead to SCD. ABS is inherited as an autosomal dominant trait and includes specific arrhythmias such as sinus node dysfunction, atrial fibrillation, long QT syndrome (LQTS) [10, 11], ventricular tachycardia (VT), idiopathic ventricular fibrillation (VF), catecholaminergic polymorphic ventricular tachycardia (CPVT), and SCD [5, 12, 13] (see Supplementary Fig. 1). Interestingly, recent studies have found that individuals carrying ANK2 gene variants could have structural heart abnormalities [14, 15, 16, 17], regardless of whether they show a prolonged corrected QT (QTc) interval. This could potentially raise their risk of developing malignant arrhythmias and SCD.
Understanding the genetic basis of these conditions is essential for developing effective treatments and interventions for managing cardiac arrhythmias and SCD. Several studies [9, 11, 12] have investigated the role and mechanisms of ANK2 gene variants in cardiovascular diseases. This review explores the relationship between ANK2 gene variants and their associated cardiac phenotypes (Table 1), summarizes recent findings on genetic mechanisms and clinical impacts, and highlights areas for future research to enhance understanding and improve therapies for cardiovascular diseases.
| Variants in ANK2 | Location | Cardiac arrhythmias | Cardiac structure abnormalities | SCA/SCD | Family history | Functional analysis | |||||||||||
| LQTS | CPVT | BrS | SND | WPW | CD | SVT/AF | VT/VF | TdP | DCM | HCM | ACM | CHD | |||||
| S646F | MBD | √ | √ | √ | √ | √ | √ | ||||||||||
| R990Q | SBD | √ | √ | √ | √ | √ | |||||||||||
| Q1316H | SBD | √ | √ | ||||||||||||||
| T1437I | SBD | √ | √ | √ | √ | ||||||||||||
| E1458G | SBD | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | ||||||
| M1988T | Exon 38 | √ | √ | √ | |||||||||||||
| V3634D | RD | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | ||||||
| L3740I | RD | √ | √ | √ | √ | √ | |||||||||||
| T3744N | RD | √ | √ | √ | √ | √ | |||||||||||
| R3906W | RD | √ | √ | √ | √ | √ | √ | √ | |||||||||
| E3931K | RD | √ | √ | √ | √ | √ | √ | √ | √ | √ | √ | ||||||
| G53A | MBD | √ | √ | ||||||||||||||
| S105T | MBD | √ | |||||||||||||||
| Y148H | MBD | √ | √ | √ | √ | ||||||||||||
| V248M | MBD | √ | √ | √ | |||||||||||||
| G290S | MBD | √ | √ | ||||||||||||||
| T466M | MBD | √ | √ | ||||||||||||||
| G475R | MBD | √ | |||||||||||||||
| Q476R | MBD | √ | √ | ||||||||||||||
| R539W | MBD | √ | √ | ||||||||||||||
| V543M | MBD | √ | √ | ||||||||||||||
| D683G | MBD | √ | √ | ||||||||||||||
| V708M | MBD | √ | |||||||||||||||
| G761S | MBD | √ | √ | √ | |||||||||||||
| L765M | MBD | √ | |||||||||||||||
| I777V | MBD | √ | |||||||||||||||
| T790I | MBD | √ | √ | ||||||||||||||
| T825I | MBD | √ | |||||||||||||||
| G859R | MBD | √ | √ | ||||||||||||||
| R895Q | MBD | √ | √ | √ | |||||||||||||
| I964V | MBD | √ | √ | ||||||||||||||
| R982Q | SBD | √ | |||||||||||||||
| L1128V | SBD | √ | √ | √ | √ | ||||||||||||
| R1305Q | SBD | √ | √ | √ | |||||||||||||
| T1437M | SBD | √ | √ | ||||||||||||||
| K1556* | SBD | √ | |||||||||||||||
| R1582Q | Exon 38 | √ | |||||||||||||||
| V1593F | Exon 38 | √ | |||||||||||||||
| R1604K | Exon 38 | √ | |||||||||||||||
| K1626E | Exon 38 | √ | |||||||||||||||
| H1806Q | Exon 38 | √ | √ | ||||||||||||||
| V1857E | Exon 38 | √ | √ | √ | √ | ||||||||||||
| G1920R | Exon 38 | √ | |||||||||||||||
| E1934V | Exon 38 | √ | |||||||||||||||
| I2050T | Exon 38 | √ | √ | ||||||||||||||
| T2059M | Exon 38 | √ | √ | √ | |||||||||||||
| R2069H | Exon 38 | √ | √ | √ | √ | ||||||||||||
| E2186K | Exon 38 | √ | √ | ||||||||||||||
| K2337E | Exon 38 | √ | √ | ||||||||||||||
| E2378K | Exon 38 | √ | √ | √ | |||||||||||||
| D2445G | Exon 38 | √ | √ | √ | √ | ||||||||||||
| R2466H | Exon 38 | √ | |||||||||||||||
| V2708A | Exon 38 | √ | |||||||||||||||
| D2802H | Exon 38 | √ | |||||||||||||||
| A2948G | Exon 38 | √ | √ | √ | |||||||||||||
| E3016K | Exon 38 | √ | √ | √ | |||||||||||||
| E3062G | Exon 38 | √ | |||||||||||||||
| D3177H | Exon 38 | √ | √ | ||||||||||||||
| T3227P | Exon 38 | √ | |||||||||||||||
| I3285T | Exon 38 | √ | √ | ||||||||||||||
| S3300R | Exon 38 | √ | |||||||||||||||
| I3437T | Exon 38 | √ | √ | ||||||||||||||
| N3554Y | Exon 38 | √ | √ | ||||||||||||||
| E3570K | RD | √ | √ | √ | |||||||||||||
| W3620R | RD | √ | √ | √ | √ | √ | √ | ||||||||||
| L3621V | RD | √ | √ | √ | |||||||||||||
| V3634I | RD | √ | √ | ||||||||||||||
| E3650Q | RD | √ | √ | ||||||||||||||
| R3659L | RD | √ | |||||||||||||||
| K3694R | RD | √ | |||||||||||||||
| E3696K | RD | √ | √ | ||||||||||||||
| V3774M | RD | √ | |||||||||||||||
| T3776A | RD | √ | √ | ||||||||||||||
| S3839T | RD | √ | √ | ||||||||||||||
| R3842W | RD | √ | √ | ||||||||||||||
| I3940R | RD | √ | √ | √ | √ | ||||||||||||
ANK2, ankyrin 2; LQTS, long QT syndrome; CPVT, catecholaminergic polymorphic ventricular tachycardia; BrS, Brugada syndrome; CD, conduction disorders; AF, atrial fibrillation; SVT, supraventricular tachycardia; WPW, Wolff Parkinson White Syndrome; SND, sinus node dysfunction; VT, ventricular tachycardia; VF, ventricular fibrillation; TdP, torsade de pointes; SCA, sudden cardiac arrest; SCD, sudden cardiac death; DCM, dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy; ACM, arrhythmogenic cardiomyopathy; CHD, congenital heart disease; MBD, membrane-binding domain; SBD, spectrin-binding domain; RD, regulatory domain.
Table 2 (Ref. [8]) presents the functional analysis of specific ANK2 gene variants. To understand the roles and mechanisms of these ANK2 variants, researchers employed a variety of methodologies and techniques. One approach evaluates the prevalence of the minor allele in different populations and examines co-segregation patterns to understand variant frequency and heritability. Protein sequence and biophysical modeling techniques are used to analyze ANK2 protein structure and predict how variants affect its function. In silico analyses further predict the effects of these variants on ankyrin-B. Furthermore, both in vitro and in vivo assays are crucial, with the latter involving the introduction of ANK2 variants into mice to observe physiological responses and assess pathogenic potential. Despite the value of in vivo murine models, few studies have used them to analyze ANK2 variants. Fig. 1 displays 11 ANK2 gene variants, primarily located in the CTD, that have undergone functional analysis.
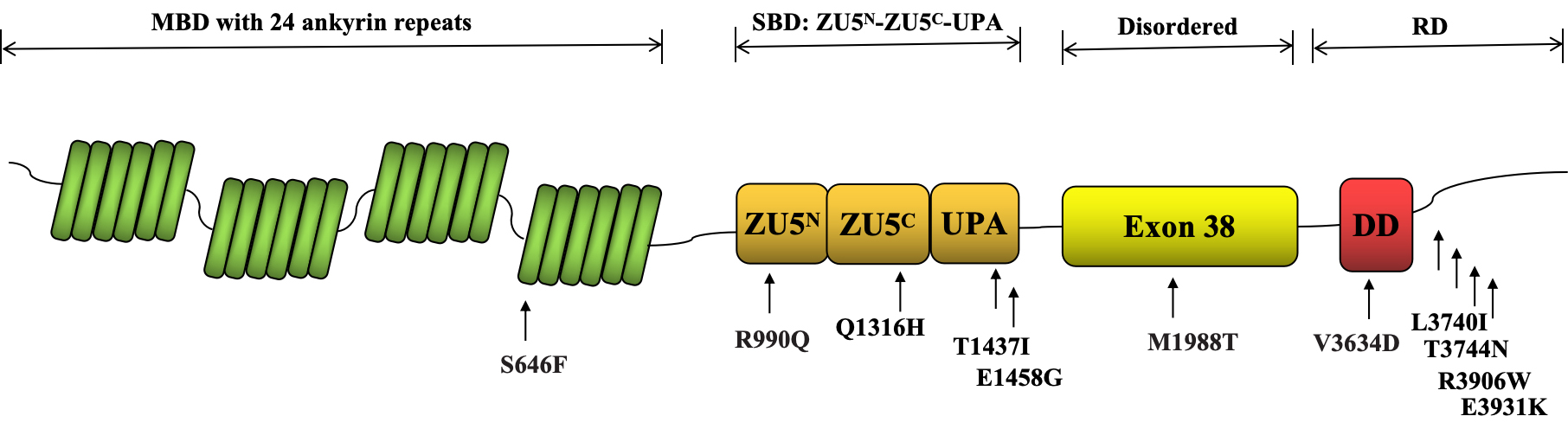 Fig. 1.
Fig. 1.
Functional Analysis of ANK2 Gene Variants Mapped to the NCBI Reference Sequence NP_001139.3. The N-terminal MBD comprises 24 ankyrin repeats, which are highly conserved and are involved in interactions between proteins. The SBD is composed of a tandem of ZU5N-ZU5C-UPA domains. The disordered region (exon 38) is a flexible linker between the SBD and the RD. The RD includes a DD and a variable CTD. MBD, membrane-binding domain; SBD, spectrin-binding domain; RD, regulatory domain; CTD, C-terminal domain; DD, death domain; ZU5N-ZU5C-UPA, Zinc-binding domain 5 N-terminal-Zinc-binding domain 5 C-terminal-UPA; NCBI, National Center for Biotechnology Information.
| ANK2 gene variants | Annotation | dbSNP | Allele frequency (gnomAD) | Functional analysis | ANK2 function | Downstream binding partners | ||||||
| * | # | ※ | In silico analysis | In vitro assays | Knock-out mice& | Knock-in mice | Expression | Localization | ||||
| S646F | S646F | S646F | Missense | rs786205724 | - | √ | √ | × | × | Reduced | Abnormal | NCX1 |
| R990Q | R990Q | R990Q | Missense | rs373261456 | 4.83251 × 10-05 | √ | √ | √ | × | NA | Abnormal | |
| Q1316H | Q1316H | Q1283H | Missense | rs755373114 | 9.91229 × 10-06 | √ | √ | × | √ | Unchanged | Unchanged | PP2A/B56 |
| T1437I | T1437I | T1404I | Missense | - | - | √ | √ | √ | × | Unchanged | Unchanged | Na+/K+ ATPase; NCX1; IP3R |
| E1458G | E1458G | E1425G | Missense | rs72544141 | 7.4501 × 10-04 | √ | √ | √ | √ | Reduced | Abnormal | Na+/K+ ATPase; NCX1; IP3R |
| M1988T | - | - | Missense | rs2154021231 | 6.20 × 10-07 | √ | √ | × | × | Reduced | Abnormal | WNT/ |
| V3634D | V1549D | V1516D | Missense | rs66785829 | 2.08879 × 10-03 | √ | √ | √ | × | Reduced | Abnormal | Na+/K+ ATPase; NCX1; IP3R |
| L3740I | L1655I | L1622I | Missense | rs35530544 | 1.753345 × 10-03 | √ | √ | √ | √ | Unchanged | Unchanged | Na+/K+ ATPase; NCX1; IP3R |
| T3744N | T1659N | T1626N | Missense | rs121912705 | 1.171034 × 10-03 | √ | √ | √ | × | Unchanged | Unchanged | Na+/K+ ATPase; NCX1; IP3R |
| R3906W | R1821W | R1788W | Missense | rs121912706 | 1.250316 × 10-03 | √ | √ | √ | × | Reduced | Abnormal | Na+/K+ ATPase; NCX1; IP3R |
| E3931K | E1846K | E1813K | Missense | rs45454496 | 03.42896 × 10-03 | √ | √ | √ | × | Unchanged | Unchanged | Na+/K+ ATPase; NCX1; IP3R |
ANK2 variants were mapped to the following reference sequences: *NP_001139.3; #NP_066187.2; and are associated with findings reported by ※Wang et al. [8] (DOI: 10.1073/pnas.1200613109).
&Introducing a mutant form of ankyrin-B into the myocytes of ANK2 haploinsufficient mice does not restore normal function, indirectly supporting that these ANK2 variants have a significant impact on the heart’s function.
gnomAD, Genome Aggregation Database (http://gnomad.broadinstitute.org); NCX1,
Na+/Ca2+ exchanger type 1; IP3R, inositol 1,4,5 trisphosphate receptor;
PP2A, protein phosphatase 2A; RyR2, ryanodine receptor type 2; dbSNP, database of single nucleotide polymorphisms; NA, not available; WNT, wingless-type mouse mammary tumor virus integration site family; B56
In terms of the pathogenic mechanisms, the majority of ANK2 variants have been found to disrupt the normal function of ankyrin-B in multiple ways. These variants affect the expression levels and cellular localization of ankyrin-B, which in turn disrupts the proper interaction with its downstream binding partners. The mechanisms behind reduced ankyrin-B expression remain unclear but are thought to involve autophagy and ubiquitin-proteasome degradation pathways [18, 19, 20].
A key partner affected by ankyrin-B dysfunction is the Na+/Ca2+
exchanger type 1 (NCX1) protein, essential for the regulation of intracellular
calcium dynamics. Ankyrin-B dysfunction can cause erratic calcium dynamics,
leading to cellular afterdepolarizations and extrasystoles that may trigger
arrhythmias. In addition to NCX1, ANK2 variants also affect other key
binding partners, including the inositol 1,4,5-trisphosphate receptor (IP3R),
Na+/K+ ATPase, protein phosphatase 2A (PP2A), and
However, the exact pathogenic mechanisms underlying ANK2-related cardiac structural abnormalities remain poorly understood. Further studies are needed to clarify how these variants contribute to structural abnormalities in the heart. This will enhance our understanding of ankyrin-B’s role in maintaining the heart’s structural and functional integrity.
The frequency of a minor allele can serve as a valuable indicator in predicting a variant’s contribution to disease. If a variant is rare or absent in large population cohorts, it may indicate a potential disease-causing variant. Table 2 shows the allele frequencies of ANK2 variants from the Genome Aggregation Database (gnomAD). Several ANK2 variants previously reported in the literature show higher allele frequencies within the gnomAD population. The associations of some ANK2 variants with diseases such as LQTS, Brugada Syndrome, and CPVT is debated over their pathogenicity, partly due to the high allele frequency of these ANK2 variants in the general population [27, 28, 29, 30, 31].
Giudicessi and Ackerman [32] conducted a comprehensive evaluation of the genetic link between established loss-of-function ANK2 variants and cardiac phenotypes in humans, which are often associated with ABS. They performed a retrospective review of medical records from 1727 patients referred to the Mayo Clinic Genetic Heart Rhythm Clinic to identify individuals carrying alleged ABS-causative ANK2 variants. These variants were mapped to the current reference transcript (NP_001139.3) and their frequencies compared with gnomAD data. Among 1727 patients, only 12 (0.7%) carried 4 ANK2 variants (p.E1458G, p.V3634D, p.T3744N, p.R3906W) linked to ABS. These variants were not significantly enriched in clinical settings and showed higher frequencies in public exomes/genomes reported by gnomAD. Further examination revealed that only a few of the 12 cases experienced cardiac events possibly related to ABS, while most remained asymptomatic. This suggests that these ANK2 variants are not likely to cause a monogenic condition predisposing an individual to SCD. Due to limited access to detailed genetic ancestry and clinical data, further genotype-phenotype studies—potentially through a multicenter registry—are needed to clarify the role of ANK2 loss-of-function in ABS.
Minor allele frequency (MAF) cut-offs, such as 1% in the general population, are often used to suggest potential pathogenicity, with lower frequencies indicating rarer and possibly more pathogenic variants. In addition, the threshold can be as high as 5% in studies considering population-specific effects. However, MAF is not a definitive measure, as gene variants with high MAF (e.g., ANK2 gene) can still be disease-associated under certain conditions. Therefore, beyond MAF, assessing pathogenicity should consider other supporting evidence such as family history and functional data.
Heterozygous ANK2 knockout murine models [9, 11], exhibiting ANK2 haploinsufficiency show dysregulated calcium levels, resulting in various complex cardiac phenotypes. These phenotypes include atrial arrhythmias, sinus bradycardia, QTc interval prolongation, catecholamine-induced ventricular arrhythmias, and an elevated risk of SCD. Similar phenotypes are observed in patients with ABS associated with ANK2 variants. The introduction of a mutant ankyrin-B into the myocytes of ANK2 haploinsufficient mice fails to restore normal cardiac function, suggesting the pathogenicity of certain ANK2 variants.
Most in vivo functional analyses of ANK2 variants, as shown in Table 2, currently rely on ANK2 haploinsufficient mice. However, these models may not fully reflect the disease risks associated with ANK2 variants in humans. Therefore, the direct effects of ANK2 variants on cardiac structure and function remain unclear and warrant further investigation. Knock-in mouse models carrying specific ANK2 variants can provide deeper insights into their influence on cardiac structure and function. These models more closely replicate the genetic alterations seen in humans, aiding in accurately defining associated disease risks and advancing the understanding of the pathophysiological mechanisms of ANK2 variants. To date, only three ANK2 variants—p.L3740I [33], p.E1458G [34], and p.Q1316H [35]—have been engineered into knock-in mouse models for functional analysis. As a result, the in vivo effects and arrhythmogenic potential of ANK2 variants remain largely unexplored, necessitating further investigation.
A previous study suggests that murine models may exaggerate the physiological impact of ANK2 loss-of-function variants observed in humans [32]. Given the species-specific differences between humans and animals, there is a need for future research using models that better reflect the human condition, particularly those incorporating disease-causing ANK2 variants. This includes using patient-derived induced pluripotent stem cells, which could provide a more accurate representation of human genetic alterations and help define associated disease risks.
Table 1 provides a comprehensive list of known ANK2 gene variants associated with cardiovascular diseases, as identified in the previously published studies (see Supplementary references). These specific variants have been mapped to the NCBI reference transcript: NP_001139.3. These ANK2 gene variants are associated with a range of cardiac arrhythmias and structural cardiac abnormalities. The distribution of these variants can be observed across different protein domains, specifically the MBD, SBD, and CTD.
Prolonged QTc intervals, whether congenital or acquired LQTS, are frequently identified in patients with ANK2 variants. The p.E1458G variant of ANK2, previously denoted as p.E1425G [11], is among the earliest variant linked to prolonged QTc intervals and was initially classified as LQTS type 4. This specific variant is found to segregate with the long-QT phenotype in 22 out of 24 individuals in the studied family, thereby indicating a significant genetic link to LQTS. Functionally, the p.E1458G variant disrupts the normal function of ankyrin-B, altering calcium signaling in cardiomyocytes and potentially precipitating extrasystoles, which may explain the arrhythmias in affected individuals. This research highlights ankyrin-B as the first non-ion channel protein implicated in inherited arrhythmias. The discovery of ankyrin-B’s role in LQTS introduces a new pathophysiological mechanism and broadens our understanding of inherited arrhythmias, with implications for targeted therapy development.
While debates continue on how ANK2 variants influence the QTc interval, it is important to note that not all individuals with ANK2 variants experience a prolonged QTc interval. Indeed, loss-of-function variants in the ANK2 gene are linked to various cardiac arrhythmias beyond LQTS. These include sinus node dysfunction [9, 11, 36, 37, 38], conduction disorders [13, 39], atrial fibrillation [9, 40], ventricular tachycardia/ventricular fibrillation, CPVT [13, 36], Wolff-Parkinson-White Syndrome (WPW) [41], Brugada Syndrome [42], and Torsade de Pointes [9, 43] (Table 1).
The mechanisms underlying the diversity of arrhythmia phenotypes associated with ANK2 variants are not fully understood and remain a focus of ongoing research. The clinical variability associated with ANK2 mutations may partly arise from complex interactions between ankyrin-B and other critical cellular binding partners essential for maintaining cellular structure and function. Notably, a single ANK2 variant can result in different arrhythmia phenotypes among affected individuals, a common occurrence in genetic diseases. The same variant does not uniformly manifest in all individuals due to varying penetrance and expressivity, influenced by modifying genes.
Indeed, ANK2 variants are often found alongside other genetic mutations in an individual’s genome [14, 15, 44]. For example, the ANK2 p.E3931K variant has been shown to exacerbate the cardiac phenotype in individuals carrying the KCNH2 p.H562R variant [44]. In isolation, the p.E3931K variant is linked to age-related conduction diseases, while individuals with only the KCNH2 p.H562R variant typically show no symptoms. This suggests that the cumulative effects of multiple genetic variants—ANK2 and KCNH2 in this case—could influence the manifestation and severity of the disease. This raises the possibility that ANK2 variants may play a key role in a complex disease etiology that could be oligogenic or polygenic. Given the complexity involved, further research is essential to understand how ANK2 variants contribute to the pathophysiology of arrhythmias.
Beyond various cardiac arrhythmias, loss-of-function variants in the
ANK2 gene are linked to structural abnormalities, such as hypertrophic
cardiomyopathy (HCM) [14], dilated cardiomyopathy (DCM) [17], arrhythmogenic
cardiomyopathy (ACM) [45], and congenital heart disease [16]. A prior study
conducted by Lopes et al. [14] investigated the genotype-phenotype
correlations in HCM by employing high-throughput sequencing to analyze genetic
variants. This study involved a thorough clinical assessment and genetic
examination of 41 inherited heart disease-associated genes across 874 unrelated
and consecutively recruited patients. The findings revealed a significant link
between rare ANK2 variants and the severity of left ventricular
hypertrophy in HCM. Specifically, carriers of these ANK2 variants
exhibited significantly greater maximum left ventricular wall thickness. These
findings suggest the potential role of ANK2 in modulating the clinical
expression of the HCM phenotype. Although direct causality between ANK2
gene variants and alterations in left ventricular morphology is not established,
ANK2 may exert an indirect effect on cellular phenotype through
interactions with proteins involved in calcium regulation and
Roberts et al. [15] reported a case of arrhythmogenic right ventricular
cardiomyopathy (ARVC) found during an autopsy of an individual with ABS. Further
genetic screening through exome sequencing identified a novel ANK2
variant (p.M1988T) in the ARVC-affected family. Subsequently, a comprehensive
genetic screen identified several rare ANK2 variants in a cohort of
patients with genotype-negative ARVC, indicating the presence of the disease
phenotype without known pathogenic variants in conventional ARVC-associated
genes. Probands with these rare ANK2 variants exhibit ventricular
arrhythmias and structural abnormalities such as biventricular dilation and
cardiac fibrosis. Functionally, while the direct functions and mechanisms of the
ANK2 variants were not explored, the researchers observed severe cardiac
abnormalities characteristic of ACM in the ANK2-cKO mouse model, a
genetically engineered model with cardiomyocyte-specific deletion of ankyrin-B.
These abnormalities primarily included biventricular dilation, reduced ejection
fraction, cardiac fibrosis, premature death, sinus bradycardia, QTc interval
prolongation, and catecholamine-induced ventricular arrhythmias. Interestingly,
despite the absence of fatty infiltration typically seen in human ACM,
ANK2-cKO mice exhibited a preserved desmosomal structure, suggesting
that ankyrin-B dysfunction may trigger ACM through a distinct pathophysiological
mechanism. This study further emphasized the disrupted interaction between
ankyrin-B and
Swayne and colleagues [16] conducted a comprehensive study on the ANK2 p.S646F variant, which was identified in two multigenerational families from the Gitxsan First Nation, a community with a high incidence of LQTS. This variant, located within the MBD of the ANK2 gene, is associated with a spectrum of cardiac phenotypes, including LQTS, WPW, congenital heart malformations, and DCM. The p.S646F variant leads to loss of ankyrin-B function, resulting in decreased expression levels and abnormal cellular localization in both cultured H9c2 cells and primary cardiomyocytes. The expression levels of ankyrin-B might be modulated by the proteasome and the p.S646F variant could impair cell viability [18]. Furthermore, ANK2 p.S646F adversely affects the proper membrane targeting of the NCX1, a key binding partner of ankyrin-B, indicating disrupted interaction between ankyrin-B and its downstream targets. This research identifies ANK2 p.S646F as a significant genetic factor in various cardiac phenotypes, emphasizing the essential role of ankyrin-B in cardiac structure and function.
While these studies illuminate the potential influence of ANK2 variants, their exact impact on structural heart diseases remains largely uncertain [17], emphasizing the urgent need for further exploration. Currently, many of these variants are classified as variants of uncertain significance, highlighting the need for ongoing research. Future studies are crucial to delineate the specific contributions of these ANK2 variants for the onset and progression of structural heart diseases, offering critical insights that could inform therapeutic strategies.
As gene sequencing technology continues to advance, a growing number of ANK2 variants, including both coding and non-coding types, are expected to be identified. While most reported variants are heterozygous missense mutations, attention should also be directed toward other mutation types, such as homozygous mutations. Further investigation is warranted to elucidate the roles of these ANK2 variants in cardiovascular diseases, particularly regarding their influence on the development and progression of structural heart diseases [15, 16]. Given that many ANK2 variants are currently classified as variants of uncertain significance, it is crucial to enhance our understanding of the pathophysiological mechanisms linking these variants to cardiac abnormalities and arrhythmias.
Although in vivo murine models have provided valuable insights, their ability to accurately represent the disease risks associated with ANK2 variants in humans may be limited. There is also evidence suggesting that these models may exaggerate the physiological impact of ANK2 loss-of-function variants in humans [32]. This underscores the necessity for research that considers species-specific differences and the development of more human-relevant models. Patient-derived induced pluripotent stem cells could better reflect human genetic alterations, facilitating the precise determination of disease risks. Future studies should also explore the potential for ANK2 variants, in combination with other genetic factors, to contribute to a complex disease etiology that could be oligogenic or polygenic, broadening our understanding of cardiovascular disease genetics. Additionally, further genotype-phenotype association studies, potentially involving multicenter registries, are essential to clarify the role of loss-of-function ANK2 variants in ABS pathogenesis.
Both canonical and noncanonical (giant) ankyrin-G variants have been identified as crucial for cardiovascular function and overall heart health [46, 47]. Canonical ankyrin-B is essential for normal cardiac function, while noncanonical giant ankyrin-B isoforms are linked to neurological disorders [48, 49, 50]. Despite this, the specific role of giant ankyrin-B in cardiovascular tissues remains largely unexplored. However, the specific role of giant ankyrin-B in cardiovascular tissues remains largely unexplored. Preliminary findings have identified certain variants within the giant ANK2 gene, particularly in exon 38 (e.g., ANK2 p.M1988T), among patients with cardiovascular diseases. However, the precise role and underlying mechanisms of these variants remain to be elucidated. The precise role and underlying mechanisms of these variants require further investigation to elucidate the function of giant ankyrin-B in cardiovascular diseases.
According to the Human Protein Atlas (https://www.proteinatlas.org), the ANK2 gene is most highly expressed in cardiomyocytes, followed by fibroblasts, endothelial cells, macrophages, and T-cells. While cardiomyocytes have been the focus of ANK2 research, the potential role of ANK2 in other myocardial cells in heart diseases is an area that requires further investigation.
Despite progress in identifying ANK2 variants and their potential implications in cardiovascular diseases, significant gaps in our understanding persist. Future research should focus on elucidating the precise mechanisms by which ANK2 variants contribute to disease. This should involve models that closely mimic human biology and examine the cumulative effects of multiple genetic variants on disease presentation and severity.
DXW and WGZ designed and revised the review. LJG has substantial contributions to the conception and design of the work and drafted the initial version of the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This work was supported by Guangdong Natural Science Foundation (2023A1515012798, 2024A1515013289); National Natural Science Foundation of China (82370383, 82100273).
The authors declare no conflict of interest. Wengen Zhu is serving as one of the Editorial Board members of this journal. We declare that Wengen Zhu had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Konstantinos P. Letsas.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/RCM26013.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.