
- Academic Editor
†These authors contributed equally.
Cerebral ischemia-reperfusion injury (CIRI) exacerbates neuronal damage through mechanisms including apoptosis and autophagy dysregulation. Resveratrol (Res), a natural polyphenol with neuroprotective properties, may alleviate CIRI-induced damage by modulating key signaling pathways. This study investigates the therapeutic effects of Res on CIRI, focusing on its role in balancing apoptosis and autophagy via regulation of nuclear receptor subfamily 3 group C member 2 (NR3C2) and tripartite motif containing 28 (TRIM28).
In vivo, cognitive impairment, neurological dysfunction, cerebral infarction, neuronal damage, and inflammatory response were assessed in Sprague Dawley (SD) rats subjected to middle cerebral artery occlusion/reperfusion (MCAO/R) using the morris water maze, Longa and Bederson scores, 2,3,5-tripheny ltetrazolium chloride (TTC) staining, hematoxylin and eosin staining, Nissl staining, and enzyme-linked immunosorbent assay (ELISA). The expression of NR3C2 and TRIM28 were analyzed by real time quantitative polymerase chain reaction (RT-qPCR) and western blot (WB). In vitro, Res effects on oxygen-glucose deprivation/reperfusion (OGD/R)-treated PC12 cells were evaluated using cell counting kit-8 (CCK-8), ELISA, terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labeling (TUNEL) staining, and WB. The relationship between NR3C2 and TRIM28 was validated using dual luciferase and chromatin immunoprecipitation followed by quantitative polymerase chain reaction (ChIP-qPCR).
Res treatment significantly improved cognitive performance in the morris water maze test, and the infarct area was reduced by 16.736%. It was accompanied by downregulation of NR3C2 and TRIM28 expression. In vitro, Res enhanced cell viability, reduced inflammatory responses and apoptosis (with a 17.70% decrease in cell apoptosis rate), and restored autophagy balance. Mechanistically, NR3C2 was shown to directly regulate TRIM28 transcription, mediating the observed neuroprotective effects.
Res inhibits NR3C2 expression, which in turn directly regulates the transcription of TRIM28 through NR3C2, alleviating apoptosis and autophagy dysregulation induced by CIRI. This mechanism clearly demonstrates the important role of NR3C2 in CIRI and reveals its regulatory relationship with TRIM28. By uncovering the neuroprotective effects of Res, we provide new insights for the treatment of CIRI and lay the foundation for future targeted therapeutic strategies involving NR3C2 and TRIM28.
Cerebral ischemia-reperfusion injury (CIRI) is a prevalent consequence of acute ischemic stroke [1]. In the acute phase of cerebral infarction, the timely restoration of blood flow is crucial for salvaging ischemic brain tissue [2]. However, the damage incurred during reperfusion can lead to additional neuronal injury and exacerbate brain dysfunction [3]. Therefore, identifying effective treatment strategies to mitigate reperfusion injury, protect neurons, and enhance patient prognosis is essential [4]. In CIRI, apoptosis and excessive represent two key pathological mechanisms. The intensified inflammatory response results in widespread neuronal apoptosis [5], leading to the destruction of neural networks and irreversible damage to brain tissue [6]. Furthermore, excessive autophagy undermines cell survival by degrading vital proteins and organelles [7]. Consequently, investigating the molecular mechanisms that influence apoptosis and autophagy in CIRI is crucial for developing therapeutic agents aimed at promoting neuronal survival, inhibiting excessive autophagy, and improving patient outcomes.
Despite progress in the understanding of CIRI mechanisms, effective treatment strategies remain limited [8]. Recent study has demonstrated that resveratrol (Res) exhibits a wide array of neuroprotective effects [9], characterized by its antioxidant and anti-inflammatory properties that mitigate neuronal apoptosis and excessive autophagy through various molecular pathways [10]. However, the specific pharmacological mechanisms in CIRI-related cell apoptosis and heightened autophagy are not yet fully elucidated.
Nuclear receptor subfamily 3 group C member 2 (NR3C2) is a nuclear receptor extensively expressed across multiple tissues, including the kidney, heart, brain, and blood vessels [11]. It regulates gene expression by binding to the promoters of target genes [12]. Recent investigations suggest that NR3C2 activation may promote apoptosis following ischemic stroke [13]. Huang et al. [14] demonstrated that inhibiting NR3C2 expression leads to reduced neuronal damage, decreased apoptosis, and diminished inflammatory responses in focal cerebral ischemia-reperfusion (FCIR) models. Nevertheless, the role of Res in neuroprotection via NR3C2 remains unclear. We hypothesize that Res may mediate apoptotic processes in CIRI by targeting NR3C2.
As an upstream transcription factor, NR3C2 is likely to regulate the transcription of downstream genes. We hypothesize that Res may influence the transcription of other genes by targeting NR3C2, thereby mediating cell apoptosis in CIRI. To identify these downstream factors, we utilized the hTFtarget database and identified tripartite motif containing 28 (TRIM28), a multifunctional protein that plays a critical role in various biological processes [15]. Existing literature indicates that knocking down TRIM28 can reduce apoptosis in myocardial ischemia-reperfusion injury [16], underscoring its significance in ischemic damage. Additionally, TRIM28 is involved in regulating the expression of autophagy-related genes [17]. However, there is currently no research addressing TRIM28’s role in apoptosis and autophagy in CIRI cells. Thus, investigating TRIM28’s involvement in Res therapy for CIRI is holds considerable importance.
This study aims to investigate the therapeutic potential of Res in CIRI, with a focus on the regulatory role of NR3C2 in TRIM28-mediated apoptosis and autophagy. The findings are expected to offer novel insights into the molecular mechanisms through which Res modulates apoptosis and autophagy in CIRI, potentially identifying new therapeutic targets.
Through Genecards database (https://www.genecards.org/) CIRI-related genes were investigated. Using Prediction database (https://prediction.charite.de/index.php), search for downstream targets of Res. Human transcription factor database (TFDB) (http://bioinfo.life.hust.edu.cn/HumanTFDB#!/) was used to query human transcription factors. On the jvenn website (https://bioinfogp.cnb.csic.es/tools/venny/index.html), perform intersection analysis on different datasets to generate Venn graphs. In the hTFtarget database (https://guolab.wchscu.cn/hTFtarget//#!/Tf), obtain downstream targets of NR3C2.
Seventy 7-week-old male Sprague-Dawley rats (weighing 230–270 g) were purchased
from Hunan SJA Laboratory Animal Co., Ltd. (Hunan, China). All rats were housed
under the following conditions: a room temperature of 25
Rats were randomly assigned to the following groups: sham, middle cerebral artery occlusion/reperfusion (MCAO/R), Res, overexpression-negative control (OE-NC), OE-NR3C2, OE-NR3C2+sh-NC, and OE-NR3C2+sh-TRIM28, with 10 rats in each group. Rats were anesthetized with 2% isoflurane (HY-A0134, Medchemexpress, Monmouth Junction, NJ, USA), and the left carotid artery, internal carotid artery (ICA), and external carotid artery (ECA) were carefully dissected. Insert a silicone-coated 3-0 monofilament nylon round head suture (Ethicon, Somerville, NJ, USA) into the ICA to close the middle cerebral artery (MCA). After 120 minutes of MCAO, the suture was removed to induce reperfusion and establish a CIRI rat model [18]. Rats in the sham group underwent the same procedure but without carotid artery ligation. All groups, except the Sham and Model groups, received intravenous injection of 30 mg/kg Res (53271ES70, Yeasen, Shanghai, China) at the start of reperfusion [19]. Rats in the OE-NC, OE-NR3C2, OE-NR3C2+sh-NC (sh-NC: 5′-TTCTCCGAACGTGTCACGTTT-3′), and OE-NR3C2+sh-TRIM28 (sh-TRIM28: 5′-ATCATGAAGGAGCTGAATAAA-3′) groups were injected with adeno-associated virus (AAV). Seven days before MCAO surgery, 4 µL of concentrated virus solution (overexpressed adeno-associated virus (VB900166-4386vrw, Vectorbuilder, Guangzhou, China), interfering adeno-associated virus (VB900185-3370auz, Vectorbuilder)) was stereotactically injected into the ipsilateral ventricle (coordinates: anterior fontanelle: anterior-posterior (AP) 1.1 mm, medial-lateral (ML) 0.8 mm, dorsal-ventral (DV) 4.2 mm). Twenty-four hours after MCAO, rats were euthanized via intraperitoneal injection of 2% pentobarbital sodium (P3761, Sigma-Aldrich, St. Louis, MO, USA) (150 mg/kg), and tissues were collected for subsequent experiments. This study was approved by the Animal Committee of Hunan Evidence-based Biotechnology Co., Ltd. (Hunan, China) (No: 202310007).
The rat PC12 cell line (MZ-2620, Mingzhoubio, Ningbo, China) was validated by short tandem repeats (STR) analysis and was cultured in DMEM (D5796, Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% FBS (HY-T1000, Medchemexpress, Monmouth Junction, NJ, USA) at 37 °C in a 5% CO2 humidified incubator until the cells reached 80% confluency [20]. Detect cells for mycoplasma contamination using mycoplasma polymerase chain reaction (PCR) Detection Kit (C0301S, Beyotime, Shanghai, China), and the result were negative. For the oxygen-glucose deprivation/reperfusion (OGD/R) treatment, the PC12 cells were first cultured in Earle’s Balanced Salt Solution (24010043, Thermo Fisher Scientific, Waltham, MA, USA) without glucose and FBS for 24 hours under hypoxic conditions (95% N2, 5% CO2, 37 °C). Subsequently, the cells were transferred to DMEM containing normal serum and cultured for an additional 24 hours. During the reoxygenation and re-glucose period, the cells were treated with Res (10 µM) for 24 hours [21].
For lentiviral infection, we utilized lentivirus containing either
overexpression of NR3C2 or a negative control, along with sh-TRIM28
(5′-ATCATGAAGGAGCTGAATAAA-3′) and its corresponding negative control
short hairpin RNA (shRNA) lentivirus (22917-1, Genechem, Shanghai, China). Cells were
inoculated at a concentration of 1
To evaluate the learning ability of rats, they were tested in a circular water pool with a diameter of 1.5 meters, and an external maze was set up in the laboratory to provide visual cues. The platform (with a diameter of 120 centimeters) was submerged 30 cm below the water surface, and the water temperature was maintained between 23 °C and 25 °C. The experiments included positioning and navigation tasks as well as space exploration trials. The positioning and navigation experiment lasted for 5 days, during which rats were placed in the first, second, and fourth quadrants of the pool, and their swimming trajectories were recorded within 120 seconds, with the escape latency defined as the time taken to find the platform. In the space exploration trials, the platform was removed, and rats were placed in the first quadrant, with data collected on the time to swim to the original platform location, the number of platform crossings, and the time spent in the target quadrant. To minimize the effects of circadian rhythm, experiments were conducted at 8 AM and 3 PM in a quiet laboratory with controlled temperature and light intensity [22]. Metrics such as escape latency, time spent in the target quadrant, and platform crossings were tracked and analyzed using ANY-maze video tracking software (DigBehv 4.1.9&1.0.29, YAN-MWMR, Yuyanbio, Shanghai, China).
The Longa scoring system and Bederson rating criteria were used to assess neurological deficits in rats. In the Longa system, 0 points indicate no deficits; 1 point reflects an inability to fully extend the left forepaw with mild deficits; 2 points signify left circling and moderate deficits; 3 points represent severe focal deficits with constant left turning; and 4 points indicate an inability to walk spontaneously and reduced consciousness. In the Bederson criteria, 0 points indicate no deficits; 1 point reflects loss of forelimb flexion; 2 points include loss of forelimb flexion with increased resistance to lateral thrust and reduced postural stability; 3 points signify unidirectional circling; 4 points represent longitudinal rolling or epileptic activity; and 5 points indicate a complete lack of spontaneous activity.
The brain tissue was sectioned into 5–6 consecutive coronal slices and placed in 1% 2,3,5-triphenyl tetrazolium chloride (TTC) buffer (298-96-4, Yeasen, Shanghai, China), followed by incubation at 37 °C for 20 minutes. After incubation, the slices were postfixed in 4% paraformaldehyde (MA0192, Meilune, Dalian, China) for 24 hours and then photographed. The red area represents the non-infarcted tissue portion, while the light white area shows the infarcted tissue portion. The morphometric analysis of the infarcted area was performed using Image (V1.8.0.112, NIH, Madison, WI, USA).
Collect rat brain tissue and fix it with 4% paraformaldehyde for 24 hours. Then the brain tissue was processed into paraffin blocks, sliced into paraffin sections of 4 µm thickness, stained with H&E dye, and observed for cell morphology under a light microscope (CX43, Olympus, Tokyo, Japan).
The preparation of tissue slices is the same as described above. The tissue was stained in 0.1% toluidine blue solution (BL1138A, biosharp, Hefei, China) for 20 minutes, washed with distilled water, and differentiated in 95% ethanol (A507050-0005, Sangon, Shanghai, China) for 15 minutes. Observe the morphological changes of Nissl bodies under an optical microscope (CX43, Olympus, Tokyo, Japan).
The terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labeling (TUNEL) assay kit (C1089, Beyotime, Shanghai, China) was employed to assess cell apoptosis. Cells were fixed in 4% paraformaldehyde for 30 minutes, followed by washing with PBS (P3813, Sigma-Aldrich, St. Louis, MO, USA). To permeabilize the cell membrane, immunostaining permeabilization solution (P0097, Beyotime, Shanghai, China) was applied for 5 minutes at room temperature, then rinsed twice with PBS. TUNEL detection solution was added to the cells, and incubation proceeded at 37 °C in the dark for 60 minutes. After three PBS washes, the cells were mounted with anti-fluorescence quenching mounting medium (HY-K1042, Medchemexpress, Monmouth Junction, NJ, USA) and visualized under a fluorescence microscope. For nuclear staining DAPI (Solarbio, Beijing, China) was applied and incubated in the dark for 10 minutes. Fluorescence intensity was quantified using ImageJ software (V1.8.0.112, NIH, Madison, WI, USA).
Enzyme linked immunosorbent assay (ELISA) kits for TNF-
Total RNA was isolated from rat brain tissue and PC12 cells from each group using a rapid RNA extraction kit (DP419, Tiangen, Beijing, China), followed by real time quantitative polymerase chain reaction (RT-qPCR) using a fluorescence quantification kit (QR0100, Sigma-Aldrich, St. Louis, MO, USA). GAPDH was used as the internal reference gene, and relative messenger RNA (mRNA) levels were calculated using the 2-ΔΔCt method (Table 1).
| Gene | Direction | Sequence (5′-3′) |
| NR3C2 | F | GGGTTTGGCTGCACTGAAAG |
| R | CACTGGGAAACTGCCAAAGC | |
| TRIM28 | F | TAAGCTGACTGAGGACAAGGC |
| R | GGCCAGCAAGAACAGGTAGT | |
| GAPDH | F | TGATGGGTGTGAACCACGAG |
| R | ATTCGAGAGAAGGGAGGGCT |
NR3C2, nuclear receptor subfamily 3 group C member 2; TRIM28, tripartite motif containing 28.
Total protein from tissues and cells was extracted using radioimmunoprecipitation assay (RIPA) lysis buffer (ab170197, Abcam, Cambridge, UK). Protein concentrations were determined using a bicinchoninic acid (BCA) protein quantification kit (P0010, Beyotime, Shanghai, China). Equal amounts of protein (10 µg) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene fluoride (PVDF) membranes using the wet transfer method (ab133411, Abcam, Cambridge, UK). Seal the membrane with 5% skim milk powder (P0216, Beyotime, Shanghai, China) for 1 hour, then incubate the membrane overnight at 4 °C with the following primary antibodies: NR3C2 (0.1 µg/mL, PA5-79761, Thermo Fisher Scientific, Waltham, MA, USA), B-cell lymphoma 2 (Bcl-2) associated X (Bax) (1:1000, ab32503, Abcam, Cambridge, UK), Bcl-2 (1:500, ab194583, Abcam, Cambridge, UK), Cleaved-caspase-3 (1:500, PA5-114687, Thermo Fisher Scientific, Waltham, MA, USA), microtubule-associated protein 1A/1B-light chain 3 (LC3) (1:500, ab62721, Abcam, Cambridge, UK), Beclin-1 (1:2000, ab207612, Abcam, Cambridge, UK), sequestosome 1/p62 (p62) (1:1000, SAB5701338, Sigma-Aldrich, St. Louis, MO, USA), TRIM28 (1:500, SAB5701273, Sigma-Aldrich, St. Louis, MO, USA), GAPDH (1:2000, ab181602, Abcam, Cambridge, UK). After washing the membrane with TBST (ZS405-3, Zomanbio, Beijing, China), it was incubated with Goat Anti-Rabbit IgG H&L secondary antibody (1:1000, ab6702, Abcam, Cambridge, UK) for 2 hours. Develop using enhanced chemiluminescence (ECL) (A38554, Thermo Fisher Scientific, Waltham, MA, USA) and analyze the bands using ImageJ software (V1.8.0.112, NIH, Madison, WI, USA).
Cell viability experiments were conducted using cell counting kit-8 (CCK-8) assay kit (C0037,
Beyotime, Shanghai, China). Inoculate PC12 cells onto a 96 well plate with a cell
count of 1
Binding sites for NR3C2 on the TRIM28 promoter were predicted using JASPAR (https://jaspar.elixir.no/search?q=HLAA&collection=CORE&tax_group=vertebrates&tax_id=9606&type=all&class=all&family=all&version=all). The top three binding sites with the highest scores were mutated. Wild-type (WT) TRIM28 promoter sequences and binding site-mutated (MUT) TRIM28 promoter sequences were cloned into the pGL3 luciferase vector (Promega, Madison, WI, USA) to construct the TRIM28-WT and TRIM28-MUT vectors, respectively. Site-directed mutagenesis to generate TRIM28-MUT was performed using a site-directed mutagenesis kit (Agilent, Roseville, CA, USA). PC12 cells were co-transfected with TRIM28-WT or TRIM28-MUT reporter vectors along with NR3C2 mimetics or NC mimetics using Lipofectamine 2000 (11668030, Thermo Fisher Scientific, Waltham, MA, USA). After 48 hours, luciferase activity was measured using the Dual-Luciferase® Reporter Assay System (Promega, Madison, WI, USA).
According to the instructions of the Simple ChIP® Enzymatic Chromatin IP Kit (9003, Cell Signaling Technology, Danvers, MA, USA), PC12 cells were treated with formaldehyde for 15 minutes. The protein-chromatin cross-linking reaction was terminated with glycine. Chromatin was digested using Micrococcus nuclease and resuspended in chromatin immunoprecipitation (ChIP) buffer. Subsequently, NR3C2 antibody (4 µL/mg of lysate, PA5-81527, Thermo Fisher Scientific, Waltham, MA, USA) or IgG antibody (SAB5600195, Sigma-Aldrich, St. Louis, MO, USA) was added, and the immunoprecipitation (IP) reaction was performed overnight at 4 °C. After washing to remove unbound chromatin and reversing the cross-links, the DNA was purified for qPCR analysis.
Statistical analyses were performed using GraphPad Prism 9.0 software
(Dotmatics, Boston, MA, USA). Student t-test was used between two
groups. For comparisons among three or more groups, one-way analysis of variance
(ANOVA) was used, followed by Tukey’s post-hoc test for pairwise group
comparisons. A p-value of
To investigate the effects of Res on cognitive behavior, brain injury, and NR3C2
in MCAO/R rats, we assessed neurological and cognitive functions alongside NR3C2
expression. The morris water maze (MWM) test revealed significant cognitive impairment in the Model
group compared to the Sham group, characterized by prolonged escape latency,
decreased platform crossings, and reduced target quadrant dwell time. In
contrast, the Res group exhibited improved cognitive function, evidenced by
shorter escape latency and increased platform crossings and target quadrant dwell
time (Fig. 1A). Neurological assessments indicated that the Longa and Bederson
scores in the Model group were elevated compared to the Sham group; however,
these scores significantly decreased in the Res-treated rats (Fig. 1B),
suggesting that Res effectively mitigates neurological dysfunction. Additionally,
TTC staining demonstrated larger cerebral infarction areas in the Model group
relative to the Sham group, with the Res group showing a notable reduction in
infarction size compared to the Model group (Fig. 1C). The expression levels of
inflammatory factors TNF-
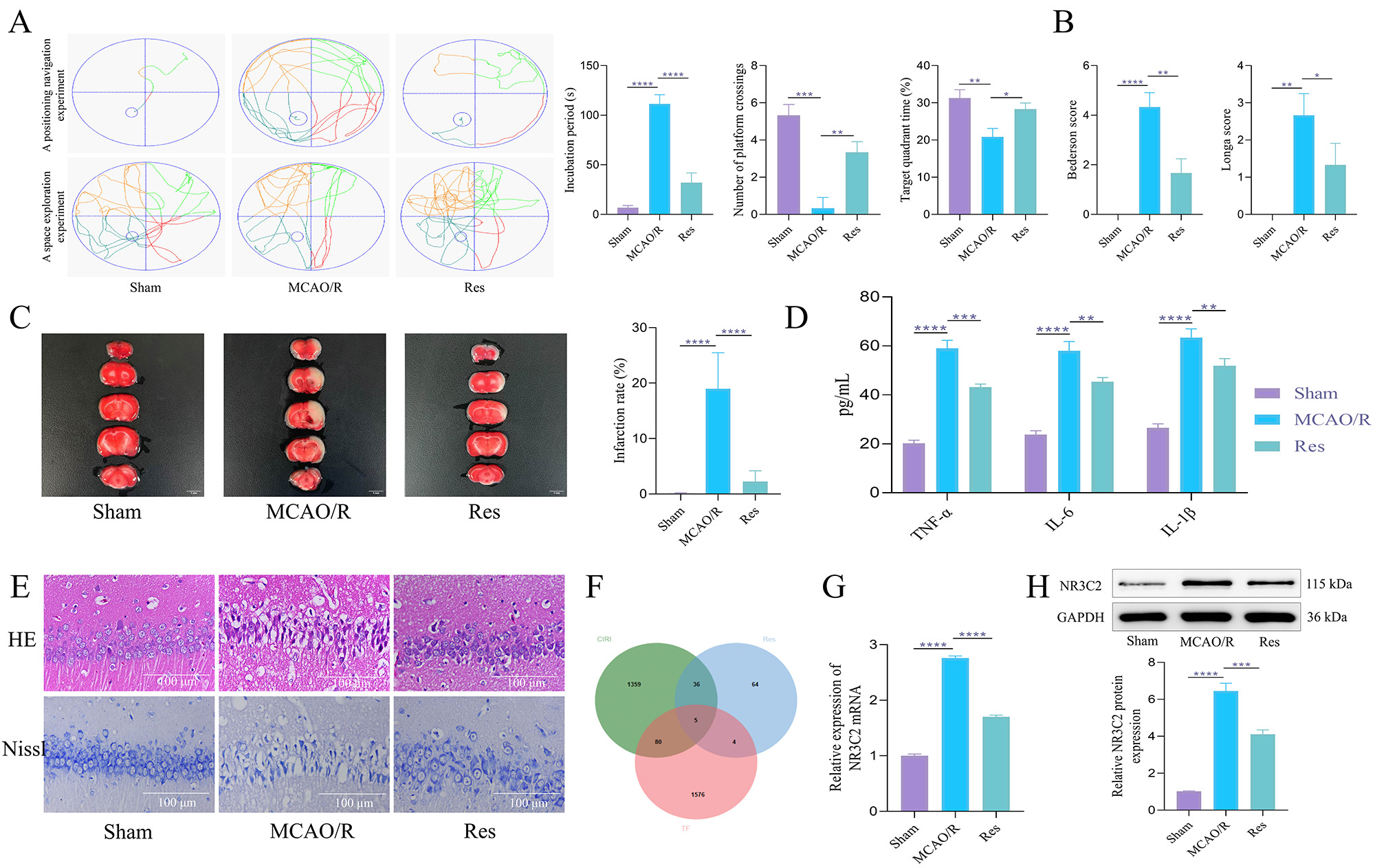 Fig. 1.
Fig. 1.
Res can alleviate nerve damage in middle cerebral artery
occlusion/reperfusion (MCAO/R) rats and downregulate nuclear receptor subfamily 3
group C member 2 (NR3C2). (A) In the Morris water maze test, the rats’
escape latency was recorded. The probe trial recorded the time spent in the goal
quadrant within 120 seconds and the number of platform crossings. N = 10. (B)
Longa and Bederson scores. N = 10. (C) 2,3,5-triphenyl tetrazolium chloride (TTC)
staining was used to evaluate the size of cerebral infarction in rats. N = 5. Scale bar = 5 mm. (D)
Enzyme linked immunosorbent assay (ELISA) was used to detect the expression of
inflammatory factors. N = 5. (E) H&E and Nissl staining were
used to observe the pathological conditions of hippocampal tissue and the changes
of Nissl bodies. N = 5. Scale bar = 100 µm. (F) Venn diagram of the
intersection of direct targets and transcription factors of Cerebral
ischemia-reperfusion injury (CIRI) and resveratrol (Res). (G) Reverse
transcription quantitative real-time PCR (RT-qPCR) was used to detect the
messenger RNA (mRNA) expression of NR3C2 in rats in each group. N = 5. (H)
Western blot (WB) was used to detect the expression of NR3C2 protein in rats in
each group. N = 5. The data was analyzed using one-way ANOVA, and the post hoc
test was conducted using Tukey’s. *p
To explore the specific mechanism by which Res protects against CIRI, we
constructed NR3C2 overexpressing cells and established in vitro model.
The results showed that compared with the Control group, the expression of NR3C2
was significantly increased (Fig. 2A,B), while cell viability was significantly
decreased (Fig. 2C) in the Model group. The expression levels of inflammatory
factors TNF-
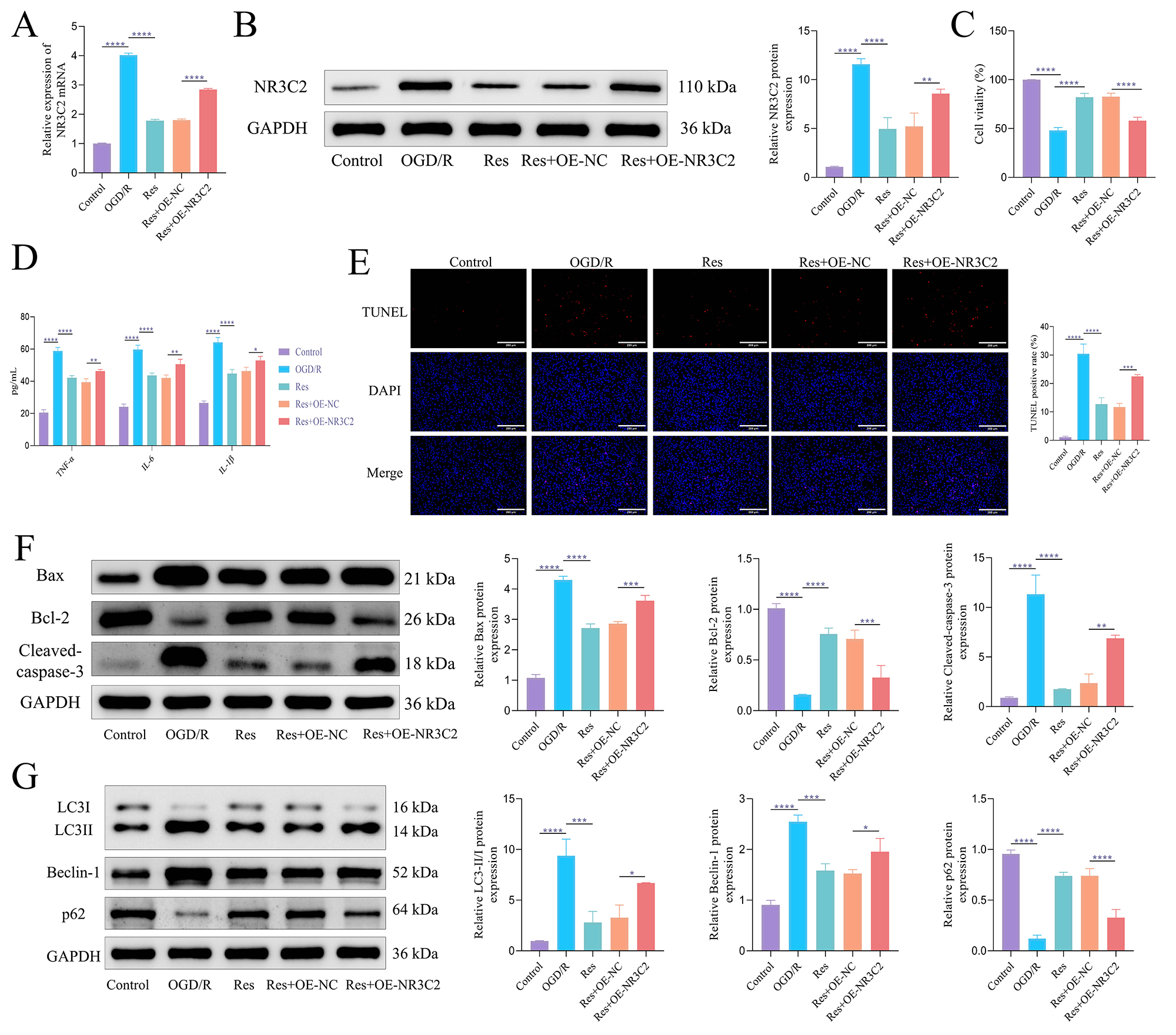 Fig. 2.
Fig. 2.
Res protects against oxygen-glucose deprivation/reperfusion
(OGD/R)-induced cell damage by inhibiting NR3C2. (A) The expression of NR3C2
mRNA was detected by RT-qPCR. (B) WB was used to detect the expression of NR3C2
protein. (C) CCK-8 was used to detect cell viability. (D) ELISA was used to
detect the content of inflammatory factors. (E) Terminal deoxynucleotidyl transferase–mediated dUTP nick end labeling
(TUNEL) was used to detect apoptosis. Scale bar = 200 µm. (F) WB
was used to detect the expression of apoptosis-related proteins. (G) WB was used
to detect the expression of autophagy-related proteins. The data was analyzed
using one-way ANOVA, and the post hoc test was conducted using Tukey’s. N = 3,
*p
To further elucidate the mechanism by which Res protects nerve cells, we identified downstream target genes of NR3C2 using the hTFtarget database and intersected these targets with CIRI-related genes via the Jvenn website. This analysis revealed nine downstream targets: peptidylprolyl isomerase A (PPIA), DNA damage-inducible transcript 3 (DDIT3), metastasis associated lung adenocarcinoma transcript 1 (MALAT1), small nucleolar RNA host gene 12 (SNHG12), alpha-enolase (ENO1), sequestosome 1 (p62/SQSTM1), TRIM28, interferon regulatory factor 9 (IRF9), and long non-coding RNA (lncRNA) THUMPD3-AS1 (Fig. 3A). Notably, while TRIM28 is known to play a role in autophagy regulation [24], its significance in the context of CIRI remains unexplored, warranting further investigation into its functional role. Our findings demonstrated that TRIM28 expression was elevated in OGD/R PC12 cells and subsequently decreased following Res treatment. However, this downregulation was inhibited by NR3C2 overexpression (Fig. 3B,C). To validate the predicted interaction between NR3C2 and TRIM28, we conducted dual luciferase assays. The results showed that NR3C2 overexpression positively regulated the wild-type TRIM28 promoter, while it had no effect on the mutant TRIM28 promoter (Fig. 3D). Additionally, ChIP quantitative PCR results revealed a significant enrichment of the TRIM28 promoter with NR3C2 compared to the IgG isotype control (Fig. 3E). These findings collectively suggest that NR3C2 regulates TRIM28 expression, positioning TRIM28 as a potential therapeutic target for Res in the treatment of CIRI.
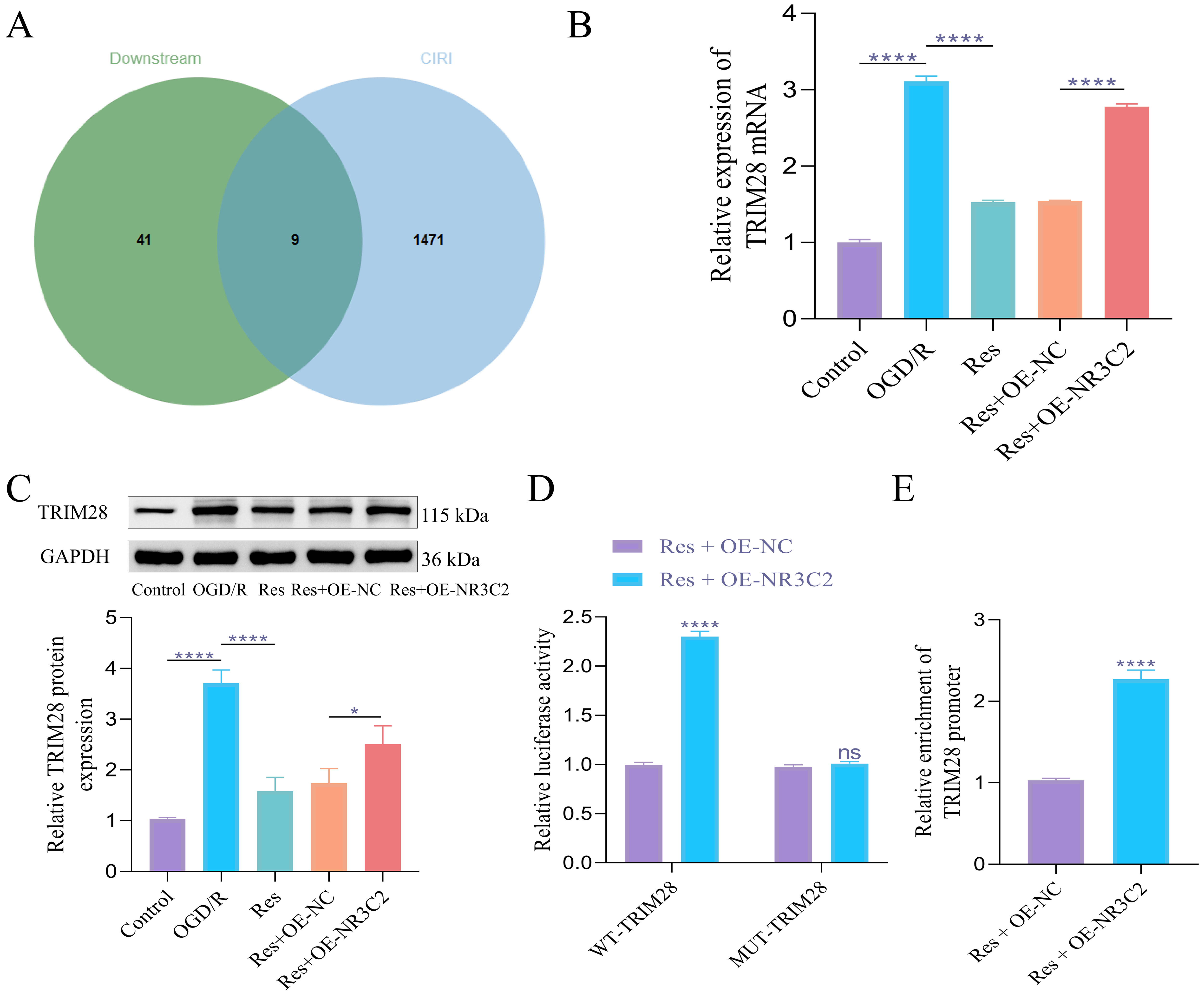 Fig. 3.
Fig. 3.
NR3C2 regulates the expression of TRIM28 in OGD/R
model. (A) Venn diagram of the intersection of NR3C2 downstream targets and
CIRI-related genes. (B) RT-qPCR was used to detect the expression of TRIM28 mRNA.
(C) WB was used to detect the expression of TRIM28 protein. (D) Dual luciferase
analysis of the regulatory relationship between NR3C2 and TRIM28. (E) ChIP qPCR
was used to detect the enrichment of the TRIM28 promoter in NR3C2. The data was
analyzed using one-way ANOVA, and the post hoc test was conducted using Tukey’s.
N = 3, ns, p
To validate the hypothesis that Res alleviates neuronal apoptosis and autophagy by inhibiting NR3C2, specifically targeting TRIM28, we conducted an in vitro experiment involving NR3C2 overexpression combined with TRIM28 knockdown. WB results showed that TRIM28 protein expression significantly decreased following knockdown (Fig. 4A), while NR3C2 protein levels remained unaffected. CCK-8 assay results indicated that cell viability increased after TRIM28 silencing (Fig. 4B). Furthermore, the expression levels of inflammatory factors were relatively reduced upon TRIM28 knockdown (Fig. 4C). Using the TUNEL assay, we observed that TRIM28 knockdown rescued apoptosis induced by NR3C2 overexpression (Fig. 4D). WB analysis was employed to evaluate protein expression levels. Results revealed that in the Res+OE-NR3C2+sh-TRIM28 group, compared to the Res+OE-NR3C2+sh-NC group, the expression of pro-apoptotic proteins Bax and cleaved caspase-3 was reduced, while Bcl-2 levels increased. Additionally, analysis of autophagy markers demonstrated a decrease in the LC3-II/LC3-I ratio and Beclin-1 expression, accompanied by an increase in P62 levels (Fig. 4E). These findings collectively suggest that Res alleviates neuronal apoptosis and excessive autophagy by directly targeting TRIM28 through the inhibition of NR3C2.
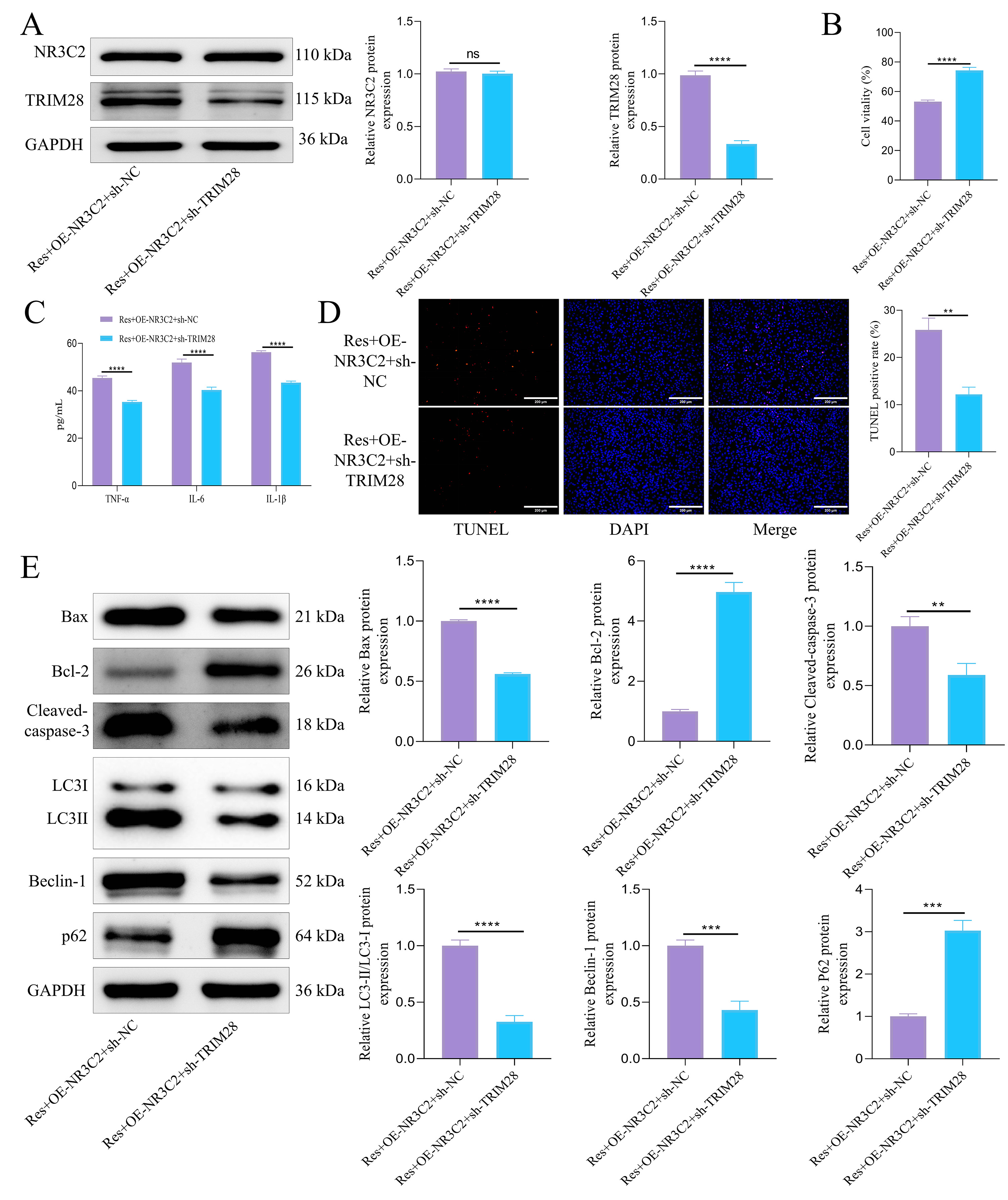 Fig. 4.
Fig. 4.
Res can protect OGD/R-induced cell damage by inhibiting NR3C2
targeting TRIM28. (A) WB was used to detect the expression of NR3C2 and TRIM28
protein. (B) CCK-8 was used to detect cell viability. (C) ELISA was used to
detect the content of inflammatory factors. (D) TUNEL was used to detect
apoptosis. Scale bar = 200 µm. (E) WB was used to detect the expression of
apoptosis-related proteins and autophagy-related proteins. Statistical analysis
was performed using student’s t-test. N = 3, ns, p
We further investigated the therapeutic effects of Res on CIRI through
in vivo experiments, specifically evaluating whether Res alleviates CIRI
by regulating NR3C2 and targeting TRIM28 expression, as well as modulating
neuronal cell apoptosis and autophagy. Our findings were consistent with those
from in vitro studies. Overexpression of NR3C2 resulted in an increase
in both NR3C2 and TRIM28 expression. In contrast, silencing TRIM28 did not lead
to significant alteration in NR3C2 levels while downregulating TRIM28 expression
(Fig. 5A). Behavioral assessments indicated that NR3C2 overexpression
significantly prolonged escape latency in rats, accompanied by a reduction in the
number of platform crossings and target quadrant dwell time. Conversely,
knockdown of TRIM28 resulted in shorter escape latency along with an increase in
both the number of platform crossings and target quadrant dwell time (Fig. 5B).
Additionally, NR3C2 overexpression was associated with elevated Longa and
Bederson scores, indicative of neurological impairment, while TRIM28 knockdown
resulted in decreased scores, although these changes were not statistically
significant (Fig. 5C). Further histopathological analysis demonstrated that NR3C2
overexpression was associated with an increased cerebral infarction area, whereas
TRIM28 knockdown led to a reduction in this area (Fig. 5D). Tissue structural
changes included a tendency towards nuclear condensation and necrosis with
diminished Nissl bodies and cell volume following NR3C2 overexpression; these
effects were reversed upon TRIM28 silencing (Fig. 5E). Moreover, NR3C2
overexpression resulted in elevated levels of inflammatory markers
TNF-
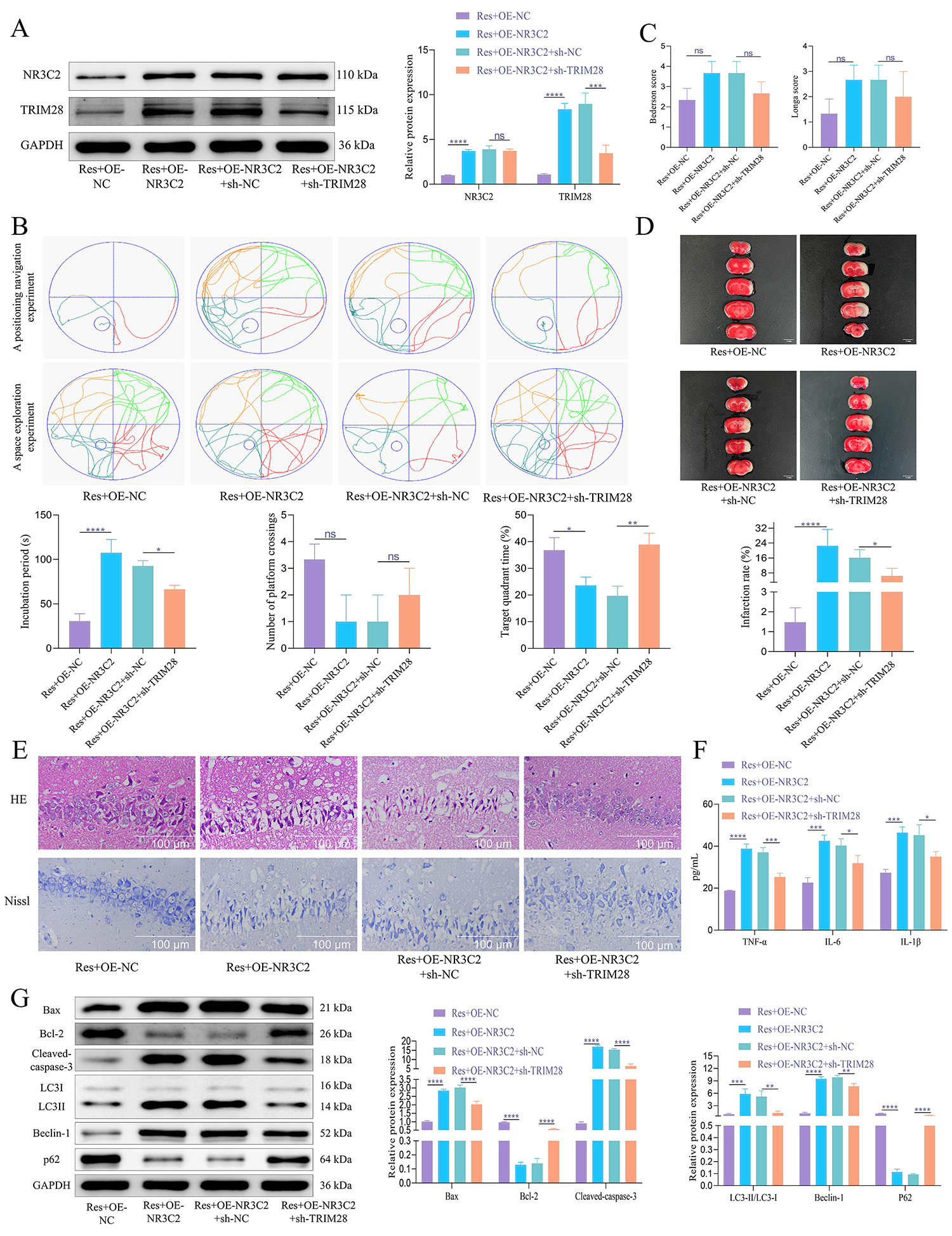 Fig. 5.
Fig. 5.
Res can alleviate neuronal apoptosis and autophagy by inhibiting
NR3C2 and reducing TRIM28 expression in MCAO/R rat brain tissue. (A) WB was used
to detect the protein expression of NR3C2 and TRIM28. N = 5. (B) In the Morris
water maze test, the rats’ escape latency was recorded. The probe trial recorded
the time spent in the goal quadrant within 120 seconds and the number of platform
crossings. N = 10. (C) Longa and Bederson scores. N = 10. (D) TTC staining was
used to evaluate the size of cerebral infarction in rats. N = 5. Scale bar = 5 mm. (E) H&E and Nissl
staining were used to observe the pathological conditions of hippocampal tissue
and the changes of Nissl bodies. N = 5. Scale bar = 100 µm. (F) ELISA was
used to detect the expression of inflammatory factors. N = 5. (G) WB was used to
detect the expression of apoptosis-related proteins and autophagy-related
proteins. N = 5. The data was analyzed using one-way ANOVA, and the post hoc test
was conducted using Tukey’s. ns, p
CIRI poses a significant clinical challenge worldwide, contributing
significantly to mortality and long-term disability. Currently, effective
therapies aimed at mitigating the effects of reperfusion-induced neuronal injury
remain limited. In recent years, Res has attracted considerable attention for its
neuroprotective properties, particularly in addressing oxidative stress,
inflammation, and apoptosis associated with various central nervous system (CNS)
disorders [25, 26]. Our findings further underscore the therapeutic potential of
Res; specifically, it demonstrates an ability to ameliorate CIRI-induced
cognitive dysfunction while attenuating both neuronal apoptosis and excessive
autophagy. This is consistent with previous studies indicating that Res mitigates
neuroinflammation and
Previous research has indicated that NR3C2 expression is tightly regulated under ischemic conditions, with its activation linked to increased apoptosis and inflammatory responses. However, the specific mechanisms by which NR3C2 mediates neuronal apoptosis in CIRI remain inadequately defined. In our study, we demonstrated a significant upregulation of NR3C2 expression in CIRI, followed by a subsequent downregulation after treatment with Res. This finding suggests that NR3C2 plays a central role in the pathological responses associated with CIRI. Furthermore, we observed that overexpression of NR3C2 diminished the neuroprotective effects of Res, indicating that NR3C2 may serve as a promising therapeutic target for intervention. Monitoring levels of NR3C2 could provide valuable prognostic information and assist in optimizing treatment strategies for patients suffering from CIRI.
Additionally, our study investigated the downstream mechanisms through which NR3C2 influences CIRI pathology. Previous research has demonstrated that NR3C2 modulates apoptosis through various downstream effectors. For instance, it can activate lipocalin 2 (LCN2) transcription, thereby promoting endoplasmic reticulum stress and apoptosis in ischemic stroke [13]. Moreover, NR3C2 regulates NLRP3 inflammasome activation, contributing to oxidative low-density lipoprotein (LDL)-induced endothelial dysfunction and apoptosis in coronary artery disease [35]. Our bioinformatics analysis identified TRIM28 as a transcriptional target of NR3C2, a relationship not previously documented within the context of CIRI. TRIM28 is known to modulate apoptosis and autophagy [36]. In this study, we report for the first time that NR3C2 binds to the promoter region of TRIM28 exerting a positive regulatory effect on its expression. Our data demonstrate that TRIM28 activation significantly amplifies neuronal apoptosis and autophagy in CIRI, exacerbating neuronal damage. Notably, treatment with Res or silencing TRIM28 resulted in reduced inflammatory markers, increased Bcl-2 expression, decreased Bax and cleaved caspase-3 levels, and altered autophagy markers, thus confirming the role of TRIM28 in these processes. These changes indicate that Res effectively inhibits cell apoptosis and reduces autophagy. Previous research has established a correlation between TRIM28 or NR3C2 and processes of cell apoptosis and autophagy. However, our study is the first to elucidate the tandem relationship between NR3C2 and TRIM28 in these processes, particularly within the context of CIRI. Given TRIM28’s involvement in various stress response pathways [37, 38, 39], further research into its regulation may be instrumental in developing effective therapeutic strategies for CIRI and other neurological disorders. Our findings highlight the importance of exploring the NR3C2-TRIM28 pathway as a potential target for CIRI treatment, providing insights into novel therapeutic strategies for managing ischemic brain injury.
In this study, we further demonstrated that Res inhibits the transcription of TRIM28 by affecting NR3C2. This finding highlights the significant potential of Res as a therapeutic agent for neurological diseases, particularly in the treatment of neuroinjury and neurodegenerative diseases (such as stroke and ischemic brain injury). However, there are some limitations in this study. First, although we observed the effects of Res on NR3C2 and TRIM28 regulation in rat in vivo and in vitro experiments, the lack of clinical CIRI samples means that our species translation lacks sufficient clinical support. Therefore, while our animal experimental results provide valuable preliminary data, further research is needed to validate these results in clinical applications. Second, the current study is limited to a rat model, and future research should expand to other animal models, or even different species, to validate the effects of Res under various physiological conditions. Furthermore, although we provide strong evidence for Res regulation of NR3C2 and TRIM28, there is still a lack of comprehensive exploration of other potential signaling pathways that may mediate its effects. Therefore, future studies should further investigate the mechanisms through which Res might exert neuroprotective effects via other pathways or molecular targets.
Res exhibits neuroprotective efficacy in CIRI by alleviating neurological dysfunction, inhibiting neuronal apoptosis, and reducing autophagy through downregulation of NR3C2 and subsequent suppression of TRIM28 transcription. This study highlights the NR3C2-TRIM28 axis as a critical pathway in CIRI pathology and supports the development of Res-based therapeutic interventions for ischemic brain injury.
The data used and analyzed during the current study are available from the corresponding author.
YL and HX contributed to the study conception and design and drafting the manuscript. Data collection and analysis and graphic design were performed by SL and ZR. SL, ZR and BW contributed to the interpretation of data for the work and revising it critically for important intellectual content. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
This study was approved by the Animal Committee of Hunan Evidence-based Biotechnology Co., Ltd. (No: 202310007). All procedures were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Not applicable.
This study is supported by Hunan Natural Science Foundation Project (2022JJ40404; 2023JJ60366).
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.