
- Academic Editor
Stroke is a major health problem with high mortality and morbidity rates, partly due to limited treatment options. Inflammation has a critical role in the secondary damage that occurs following a stroke event. Neutrophil extracellular traps (NETs) are released by neutrophils and contribute to the progression of neuroinflammation that further worsens brain damage. The prevention of NET formation at sites of brain damage has been reported to prevent neuroinflammation and improve neurological deficits. The aim of this article was to assess the importance of NETs as a treatment target for hemorrhagic stroke in light of the available evidence. NETs are network structures that consist of decondensed DNA strands coated with granule proteins such as citrullinated histones, neutrophile esterase (NE), myeloperoxidase (MPO), and high mobility group protein B1 (HMGB1). Peptidyl arginine deiminase type-IV (PAD4) plays a key role in the formation of NETs. Inhibitors of NET formation, such as the PAD4-specific inhibitor GSK484, are effective at preventing inflammation and thus ultimately reducing brain damage after stroke. In conclusion, inhibition of NETs offers a potential therapeutic strategy for hemorrhagic stroke, although further research is needed to clarify the role of NETs in this condition.
Hemorrhagic stroke (HS) is a serious condition that occurs due to bleeding into the brain after the rupture of a brain vessel [1]. About 20% of strokes are hemorrhagic, with subarachnoid hemorrhage (SAH) and intracerebral hemorrhage (ICH) each accounting for approximately 10% [2]. HS has important health, social and economic impacts, with high mortality and serious morbidity rates [3, 4].
The primary damage that occurs after hemorrhage is because of the mass effect due to the hemorrhage [5, 6]. Treatment is primarily aimed at relieving headaches and preventing complications, as well as reducing intracranial pressure and rebleeding [2, 7]. The secondary damage following HS occurs mainly after the neuroinflammatory cascade is triggered and the coagulation component is released [8, 9]. The exact nature of HS resulting from either ICH or SAH has not yet been fully elucidated. However, the basic pathophysiological mechanisms are known to be immunologically induced, and hence the search for therapeutic targets in the treatment of HS has become a very active area of research [6, 10].
Patients who survive HS develop ischemic complications resulting in neuronal damage/death. This process is thought to occur in two stages: early and delayed brain ischemia. Early brain ischemia results from toxic processes caused by transient cerebral ischemia and hemorrhage, whereas delayed brain ischemia occurs at a later stage as a result of intracranial vascular vasospasm, micro-thrombosis, and proinflammatory processes triggered by early ischemia [5, 11]. Additionally, hematoma occurs after brain injury and triggers inflammatory reactions. Among the immune cells, microglia residing in the brain are activated first and then induce peripheral immune cells, including neutrophils, monocytes and macrophages [10, 12].
Neutrophils are the most abundant cells of the immune system and are the first peripheral cells to be activated after HS [13]. The production of neutrophils increases 10-fold in the event of an external stimulus, whereupon they are recruited to the infected or damaged tissue [14]. Although microbial-induced inflammatory responses are crucial for host defense and tissue healing, they can also cause important tissue injury during a sterile inflammation, especially when there is over-reaction [6]. Recently, neutrophil extracellular traps (NETs) were reported to participate in neutrophil-mediated damage [6]. The importance of NETs is now being recognized, as they have been observed to worsen cerebral hemorrhage, neuronal damage, and disruption of the blood brain barrier (BBB) [6, 15].
The aim of this study was to assess the potential importance of NETs as a novel therapeutic approach for HS based on current evidence in the literature. Additionally, to more clearly understand the importance of this treatment strategy, we describe the possible molecular mechanisms underlying the relationship between NETs and HS.
Under normal conditions, peripheral immune cells including neutrophils cannot cross the BBB [16]. However, following events such as stroke and traumatic brain injury, large numbers of neutrophils migrate into the brain tissue as a consequence of the damaged BBB [14]. Overexpression of selectins and of some chemokines further facilitates the migration of immune cells [3, 17].
Proinflammatory cytokines [(e.g., tumor necrosis factor (TNF)
-
Neutrophils have a lifespan of less than 8 hours, as they are phagocytized and rapidly removed by macrophages after degranulation [18]. However, they are maintained in a dynamic balance between development, migration to tissues, and death. Approximately 1011 neutrophils are produced in normal adults each day [14].
Although neutrophils and NETs are associated with tissue damage, they are a double-edged sword since they also play a vital defensive role in a variety of diseases. Following tissue damage, an intricate biological process that includes the immune system is activated to reduce excessive blood and fluid loss, clear dead and devitalized tissue, and prevent infection. Circulating neutrophils are the first immune cells recruited to the injured tissue [20]. The main functions of neutrophils are phagocytosis, degranulation, and the production of ROS [15]. Besides the traditional defense mechanisms, NETs help neutrophils to immobilize and capture pathogens, thus contributing to host defense. NETs also destroy pathogens directly through the action of proteolytic enzymes such as neutrophile esterase (NE) and myeloperoxidase (MPO) located within their structures [21]. They also play an important role in immune homeostasis by bringing nutrients, immune cells, and oxygen to the wound side through their effect on angiogenesis [20]. Vital NETs are likely to show this behavior first. After releasing NETs, neutrophils survive and continue to perform other functions as part of the host response, including chemotaxis, phagocytosis, and bacterial killing [22].
NETs are network structures composed of decondensed DNA strands coated with granule proteins. They are released by neutrophils as part of their first line of defense in response to infectious and sterile stimuli [20, 21]. NET-associated material originates predominantly from the nucleus of neutrophils. It is therefore rich in core histones, but also contains high levels of proteins including citrullinated histones, NE, MPO, high mobility group protein B1 (HMGB1), peptidoglycan-binding protein, lactoferrin, pentraxin 3, serine protease, MMP-9 and proteinase 3 (PR3) [14, 23].
The formation of NETs (also called NETosis) is activated by a wide variety of
stimuli, including bacteria, phorbol 12-myristate 13-acetate (PMA), and
proinflammatory mediators such as interleukins (e.g., IL-8 and IL-1
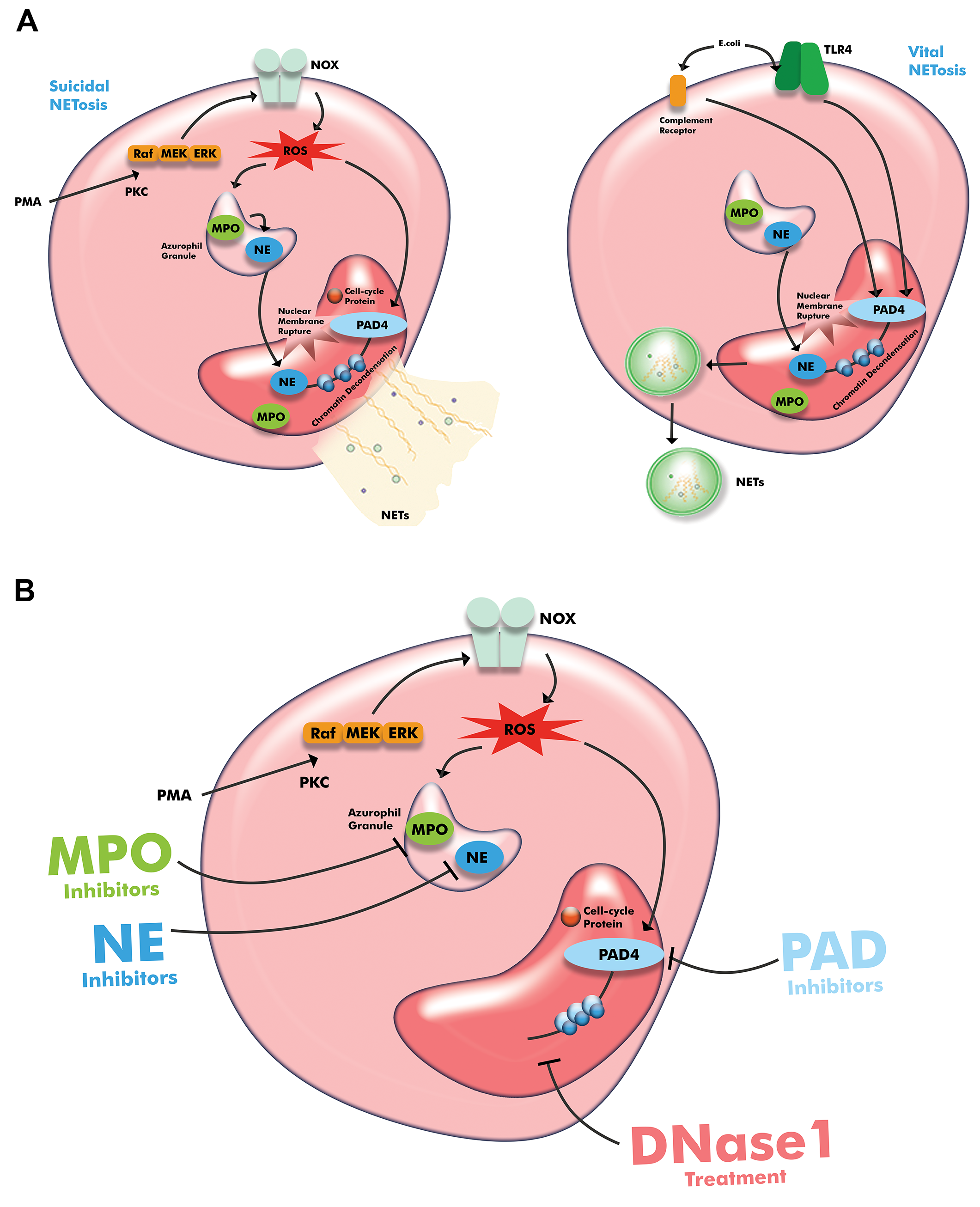 Fig. 1.
Fig. 1.
Schematic illustration of neutrophil extracellular traps (A) and therapeutic intervention (B). Drawn with Adobe Creative Suite Package (Illustrator, version 28.7.1 and Photoshop, version 25.12, Adobe Systems Incorporated, San Jose, CA, USA). Abbreviations: PMA, phorbol-12-myristate-13-acetate; PKC, protein kinase C; NOX, NADPH-oxidase; TLR, Toll-like receptor; NE, neutrophile esterase; MPO, myeloperoxidase; PAD4, peptidyl arginine deiminase type-IV; ROS, reactive oxygen species; NETs, neutrophil extracellular traps; Raf, rapidly accelerated fibrosarcoma kinase; MEK, mitogen-activated protein kinase/extracellular signal-regulated kinase; ERK, extracellular signal-regulated kinase; DNase1, deoxyribonuclease 1.
In the NOX-dependent pathway, stimuli such as lipopolysaccharide, PMA, and
TNF-
In the NOX-independent pathway, stimuli such as microbial toxins can trigger NETosis via cell membrane ligands such as TLR2 or 4, or via the complement system. In this type of NETosis, PAD4 is activated independently of ROS and the Raf-MEK-ERK pathway, and acts to decondense chromatin. NETs are released into the extracellular compartment within vesicles, thereby preserving the integrity of the plasma membrane, while the neutrophils survive and continue with their other functions such as phagocytosis [14, 29]. The type of NETosis in which neutrophils remain alive is called vital (or non-lytic) NETosis [28].
Unlike other family members, PAD4 has the unique feature of being localized in the nucleus. When the neutrophil is activated, it converts specific arginine residues in histone tails to citrulline, resulting in chromatin decondensation (in physiological conditions, neutrophil chromatin is in a condensed state) and NET formation [30]. Interestingly, neutrophils without nuclei remain morphologically intact [14]. Chromatin decondensation is thus a fundamental process in NET formation and is dependent on the presence of PAD4. NE and MPO also translocate from cytoplasmic granules to the nucleus and contribute to histone degradation and chromatin decondensation in both NOX-dependent and -independent pathways [28, 31]. While many components of the process are similar for both types of NETosis, there are fundamental differences in the stimuli, timing, and outcomes [22, 32].
The NETosis process is unique to neutrophils, and NETs normally act as a physical barrier to prevent pathogens from migrating to other areas. Hence, NETs prevent the spread of pathogens by trapping and eliminating them [33, 34]. In contrast to this beneficial role, excessive secretion of NETs causes tissue damage due to their cytotoxic and immunogenic contents, including cell-free DNA (cfDNA), MPO, and NE [33]. Because of their self-antigen structure, NETs can also potentially trigger autoimmune events [22]. These dichotomous effects of NETs are an interesting and puzzling research topic [24, 32, 35].
High levels of NETs have been associated with unfavorable outcomes in patients with SAH and ICH [36]. Zeng et al. [6] reported that the expression of NETs in the peripheral blood increased significantly 12 hours after SAH, and in the brain tissue 24 hours after SAH. Puy et al. [37] detected both neutrophil infiltration and NETs within and around hematomas in the autopsy tissue of patients who died of ICH. Neutrophils and NETs can also be considered as markers of stroke and treatment failure [36].
Neuroinflammation and micro-thrombosis are major pathophysiological processes that occur following brain injuries and are closely associated with poor prognosis [6, 38]. In brain injuries, thrombosis occurs in the damaged area and a strong inflammatory response begins simultaneously. HMGB1 derived from necrotic neurons has a damage-associated molecular pattern (DAMP) -like function in activating neutrophils, a vital component of innate immunity, via CXC chemokine receptor 4 (CXCR4)-chemokine receptor and Toll-like receptor 4 (TLR4) [39]. Activated platelets also release HMGB1, ultimately leading to further increases in thrombosis and NETosis [40, 41]. NETs serve as a structural framework for red blood cells, platelets, and adhesion molecules, resulting in a procoagulant state [36]. These findings indicate that HMGB1 plays an essential role in the link between NETosis and thrombosis [42].
In any vascular injury, NETs potentiate endothelial activation and damage. Higher level NETs are also associated with increased levels of vascular injury markers such as E-selectin and vascular cell adhesion protein-1. At the same time, activated endothelium can trigger neutrophil activation and NETosis, thereby creating a vicious cycle [36].
In vitro experiments have shown that erythrocyte-derived heme can trigger neutrophil activation and increase the release of NETs [43, 44]. Tan et al. [44] reported that disruption of NETs by DNase1 promoted tPA-induced hemo-fibrinolysis. This led to improved functional outcomes by reducing brain edema and neuron death. In another study, degradation of NETs with DNase I or inhibition of NET production by PAD4 deficiency restored tPA-induced loss of BBB integrity, thereby reducing tPA-associated cerebral hemorrhage after ischemic stroke [19]. Together, the results demonstrate that NETs play an important role in regulating tPA-induced breakdown of the BBB and in cerebral hemorrhage [19, 45].
During SAH events, marked neutrophil infiltration into the brain parenchyma is
observed together with the expression of NETs, correlating with the severity of
SAH [14]. NETs also induce the differentiation of microglia into pro-inflammatory
subtypes, causing increased production of pro-inflammatory cytokines such as
TNF-
Although the neuroinflammatory reaction has a crucial role in secondary brain damage after stroke, general immunosuppressive strategies or basic anti-inflammatory drugs fail to improve clinical outcomes [18]. Molecular models of the possible pathophysiological mechanisms are emerging as research topics, and may help to identify therapeutic targets for the prevention of neuroinflammation and early micro-thrombosis [38]. For these reasons, various immunotherapy approaches such as inactivation of immune cells that secrete inflammatory factors, inhibition of inflammatory substances, and blockade of pathways involved in the migration of immune cells to the injury site have been active areas of research [48]. Selective immunomodulatory approaches, particularly those that weaken the pro-inflammatory response and enhance the anti-inflammatory response, have become popular research topics. These include inhibiting destructive molecules, modulating the inflammatory M1 subtype of macrophages to the anti-inflammatory M2 subtype, and modulating the inflammatory N2 subtype of neutrophils to the anti-inflammatory N2 subtype [13, 48]. Other strategies include preventing or regulating the recruitment, adhesion, and migration of immune cells [18].
Shortly after different pathological events in the central nervous system,
including HS, neutrophils infiltrate the injured side and release destructive
substances that further exacerbate the damage [6, 13]. NET formation is unique to
neutrophils and is associated with adverse outcomes, including BBB leakage and
brain tissue injury [6]. Hanhai et al. [49] found that NETs induce
microglia to transform into pro-inflammatory subtypes, leading to increased
TNF-
The inhibition of some adhesion molecules, such as CXCR1/CXCR2, intercellular adhesion molecule-1 (ICMA-1), Mac-1, and P-/E-selectins, can limit neutrophil accumulation and consequently improve BBB leakage and reduce ischemic damage in animal models [50, 51, 52]. Through the peripheral application of CXC chemokine ligand 1 (CXCL1), Stamatovic et al. [48] were able to direct neutrophils from the damaged area to the periphery, thereby improving lesions caused by neutrophil-mediated damage. In addition to significantly reducing neuroinflammation and infarct size, these authors also reported a lower mortality rate compared to the control group.
Neutrophil depletion attenuates tissue inflammation, partly through the reduction of NET formation, and can even reverse cerebral vasoconstriction in SAH models [6, 53]. Blocking the formation of NETs by targeting neutrophils provides indirect evidence for a link between early brain injury and NETs [6], and may be considered an optimal treatment strategy.
In addition to blocking neutrophil recruitment and activation, other targeted therapeutic strategies are inhibition of NET formation, and promotion of NET degradation [54, 55]. Targeted drugs that inhibit hua4, HMGB1, NADPH oxidase and ROS production all suppress NET formation, while drugs that target degradation of the DNA backbone, histone, MPO and NE can facilitate NET degradation [54].
Activated platelets are a well-known inducer of NETosis, while NETs can in turn promote thrombosis by creating a procoagulant state [23, 36, 40]. Therefore, strategies that block this virtuous cycle by preventing essential adhesive interactions may reduce NET-mediated thrombosis. Inhibition of P- and E-selectin not only reduces neutrophil recruitment but also reduces the release of NETs by activating platelets via blockade of P-selectin glycoprotein ligand-1 (PSGL-1) [55]. Zhou et al. [38] observed that early micro-thrombosis and NETs co-localized in mice following SAH. Furthermore, neutrophil depletion and DNase1 clearance of NETs could reduce cortical micro-thrombosis and brain injury, reduce cerebral edema, and improve neurological function. Another study conducted using a rat model of intracerebral hemorrhage and intraventricular hemorrhage reported that numerous NETs formed in and around the hematoma, and that application of tPA combined with DNase1 accelerated clearance of the hematoma and reduced hydrocephalus [44]. However, the presence of NETs weakens the therapeutic effect of tPA in ischemic and hemorrhagic lesions [43, 56].
Dismantling of the NET scaffold using DNase I is one of the classical interventions. The use of DNase I mechanically disrupts the structure of NETs, which act as a scaffold for the adhesion of platelets and erythrocytes, thus inhibiting platelet aggregation and fibrin deposition [22]. Zeng et al. [6] reported on the potential role of DNase I as a therapeutic target in the prevention of early brain injury in SAH.
PAD4 plays a key role in NET formation in both the NOX-dependent and -independent pathways [6, 26] and has therefore been the subject of extensive preclinical investigation as a therapeutic target [57]. PAD4-specific inhibitors such as GSK484 and YW4-03, and non-PAD4-specific inhibitors such as Cl-amidine (N-[(1S)-1-(aminocarbonyl)-4-[(2-chloro-1-iminoethyl) amino] butyl]-benzamide, monohydrochloride), have reduced the ischemic damage in some clinical situations [6, 39, 45]. Cl-amidine is a covalent, irreversible, and potent pan-PAD inhibitor that reduces citrullinated histone H3 (citH3) and NET levels in various disease models [58]. F-amidine is a similar pan-PAD inhibitor, but is less potent [59]. Thr-Asp-F-amidine (TDFA) is a selective, irreversible inhibitor of PAD4 that positively correlates with recovery from acute lung injury in mice [60].
Pharmacological as well as genetically-related inhibition of NETosis is effective in preventing ischemia and brain damage [16]. PAD4-/- mouse neutrophils were reportedly unable to produce NETs [61]. Kang et al. [27] found there was no significant difference in ischemic lesions between WT and PAD4-/- mice at day 14, suggesting the effects of PAD4 deficiency on long-term outcomes were not secondary to reduced lesion size. Selected examples of studies that target NETosis are presented in Table 1 (Ref. [6, 25, 27, 39, 47, 50, 52, 59, 61, 62, 63, 64, 65, 66].
| Target | Therapeutic compound/strategy | Major action | Reference |
| PAD4 | GSK484 | Specific inhibition of PAD4 | [6, 61] |
| Cl-amidine | Pan-PAD inhibition | [39, 62] | |
| F-amidine | Pan-PAD inhibition | [59] | |
| PAD4 gene knockout | Decreased PAD4 expression | [27] | |
| RNase A | Decreased PAD4 expression | [63] | |
| nNIF | Inhibition of NET formation | [64] | |
| DNA | DNAase1 | DNA degradation | [6, 65] |
| NE | Sivelastat | NE inhibition | [25] |
| TREM1 | P17 | TREM1 inhibition | [47] |
| Neutrophil depletion | Adhesion molecule inhibitor (against CXCR1/CXCR2, ICAM-1, P-selectin etc.) | Reduced neutrophil count | [50, 52, 66] |
| Anti-Ly6G antibody | Reduced neutrophil count | [6, 27] |
Cl-amidine, N-[(1S)-1-(aminocarbonyl)-4-[(2-chloro-1-iminoethyl) amino] butyl]-benzamide, monohydrochloride; F-amidine, N-[(1S)-1-(aminocarbonyl)-4-[(2-fluoro-1-iminoethyl) amino] butyl]-benzamide, 2,2,2-trifluoroacetate; nNIF, neonatal NET inhibitory factor; TREM1, triggering receptor 1; CXCR1/CXCR2, CXCR1/CXCR2 chemokine receptor; ICAM-1, intercellular adhesion molecule-1; P-selectin, platelet selectin.
A reciprocal modulatory relationship exists between PAD4 and HMGB1. HMGB1 upregulates PAD4 expression in neutrophils, while PAD4 citrullinates HMGB1, leading to efficient chromatin decondensation. HMGB1 thus appears to occupy a crucial position between NETosis and thrombosis, and mediates the aggravation cascade that occurs due to PAD4/HMGB1 crosstalk [42]. In experimental models, treatment with neutralizing HMGB1 antibody or antagonistic peptide for HMGB1 suppressed NET formation [39]. However, this is constrained by the fact that the released histones and multiple proteases are not completely removed [38].
Neonatal NET inhibitory factor (nNIF) is an endogenous NET regulator found in umbilical cord blood [64]. In one study, nNIF treatment was effective at preventing platelet-derived NET formation in both human and mouse neutrophils [40]. Importantly, this effect was observed as early as 1 hour after stroke onset and provided long-term protection from the stroke. nNIF effectively inhibits the formation of NETs, does not affect the recruitment of neutrophils after stroke, and shows good therapeutic effects in animal models [20].
NE and MPO have also been investigated for their effects on NET formation. While both have been identified to play a role in NET formation, MPO has generally been considered only as a marker of NETs [31].
Zeng et al. [6] reported that administration of GSK-484, a specific
inhibitor of PAD4, reduced early brain injury after SAH, including cerebral edema
and neuronal deficit. They also found that inhibition of NETs by DNAse I
treatment or neutrophil depletion yielded beneficial results. In all three of
these approaches, pro-inflammatory cytokines, including TNF-
Wu et al. [47] reported that treatment with LP17, a TREM1 inhibitory
peptide, improved neurological performance and reduced brain water content and
neuronal damage. They also found that LP17 treatment prevented the activation and
pro-inflammatory subtype switching of microglia, consequently reducing
proinflammatory cytokines such as TNF-
Because the release process of NETs involves several elementary reactions, a wide variety of compounds could theoretically interfere with the formation of NETs [20]. The success of their preclinical studies has encouraged the use of therapeutics that target NETs. Despite some encouraging progress towards clinical translation, human studies that target neutrophils in HS are still few and unsatisfactory, and further research and clinical trials are needed.
Several properties of therapeutic reagents can limit their effectiveness, including short half-life, poor solubility, low bioavailability, and metabolic toxicity. In an attempt to overcome these constraints and improve the targeting of inflammation sites, researchers have studied neutrophils/neutrophil-derived cell membranes, as well as carriers such as nanoparticles conjugated with a selective binding peptide and targeted inhibitor (mainly PAD4 inhibitor). Mu et al. and Li et al. [16, 25] reported that nanoparticle carriers can effectively target the injury site and regulate post-injury inflammatory responses by reducing pro-inflammatory cytokine levels. The targeted delivery of therapeutic agents also plays a vital role in reducing the risk of undesirable and uncontrollable off-target systemic events [25]. One way to overcome such adverse events is to apply an external magnet so that magnetic nanoparticles loaded with functional molecules are localized to the injured area. The multifunctional design of magnetic nanoparticles [67] could also be used as a multimodal theranostic platform for stroke diagnosis, monitoring and treatment.
Biomarkers play a critical role in basic and clinical research, as well as in clinical practice [22, 54]. Stroke is a major public health problem that causes death and serious disability. Standardized biomarkers may therefore be used to help diagnose stroke and to monitor its severity and prognosis [36].
In addition to an increased absolute neutrophil count and neutrophil-to-lymphocyte ratio, elevated NET levels in both peripheral blood and intracranial tissue are strongly associated with stroke severity and adverse outcomes. Increased levels of specific molecules in the peripheral blood have also been proposed as markers of NETosis, including MPO-DNA, citH3, cf DNA, nucleosomes, and deoxyribonuclease I (DNAse-I) [36, 65, 68]. An increased level of markers for NET formation, such as H3cit and MPO-DNA, was also positively associated with stroke outcomes [69]. Sheng and Cui [70] performed an analysis using Mendelian randomization techniques based on genome-wide association study. Genes associated with NETs, such as autophagy-related 7 (ATG7), matrix metalloproteinase-9 (MMP9), IL-6, (DNase1), and phosphodiesterase-4B (PDE4B) were found to have important diagnostic value in the development and progression of myocardial infarction. Genetic biomarkers for stroke-related NETs may also be identified in future studies.
The highly complex and dual role of the inflammatory process is the major factor causing secondary injuries after stroke. This process complicates the clinical outcomes and therapeutic intervention following HS [2]. It is important to note that the critical role of NETs has been confirmed in clinical as well as experimental studies [37, 44]. The current evidence is still insufficient to guide recommended targeted therapy options for HS. Immunomodulatory approaches show great therapeutic potential, but a number of challenges remain.
Several of the trial agents have multifaceted therapeutic features and carry risks such as suppressing certain beneficial aspects of the immune response and causing adverse effects [68]. The temporal properties of cellular and other factors that play a role in the pathophysiological process may also be different. For example, the agent may show pro-inflammatory properties in the early period, and anti-inflammatory properties in the late period [8, 10]. Future research is likely to focus on regulating immune pathways through more targeted therapies. With additional research data, the development of well-established therapeutic dosage and standardized treatment protocols may lead to improved outcomes. Personalized approaches that take into account the clinical characteristics of the patient may also be a key strategy. NET biomarkers such as cf-DNA, nucleosomes, MPO, CitH3 or NE may be useful in this regard. Combination therapies may also be an important option due to their synergistic effects and lower dosage requirements, thus further improving outcomes and reducing off-label effects.
HS is an important clinical condition with high mortality, poor prognosis, and lack of effective treatment options. The inflammatory response is central to the poor outcomes experienced after the onset of hemorrhage. Activated neutrophils and higher levels of NETs released by neutrophils are associated with the poor outcomes after stroke. Deficiencies in the clearing and breaking down of NET residues cause tissue damage. NET inhibition significantly improves clinical outcomes after stroke. PAD4 plays a central role in NET formation, and PAD inhibitors can improve outcomes. Consequently, NET inhibition is a potential therapeutic strategy in HS. High NET levels may also serve as a marker of stroke severity, functional outcomes, and prognosis. However, most of the current literature has studied PAD inhibitors in animal models, and hence the generalizability of the inhibitory responses to humans is difficult to predict. Moreover, although similar mechanisms may be involved in conditions other than stroke, research studies on stroke are still scarce. Importantly, the uncertainty regarding the efficacy and side effects of these inhibitors in humans requires further research to elucidate the potential clinical role of NET inhibition in the treatment of HS. In order for NET-targeted therapy to be implemented in the clinic, the underlying mechanism still needs further clarification.
The article contains all supporting data.
RD and NA co-designed and conceptualized the study. RD gathered the data and NA wrote the manuscript. RD and NA read and approved the final version. Both authors contributed to editorial changes in the manuscript. Both authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
NA is voluntary consultant for Med-International UK Health Agency Ltd. The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.