

 , Rongjing Shen 1, Mengting Qian 1, Zhengjun Zhou 2, Bingqing Xie 3, Yong Jiang 3, Yang Yu 1,4,*
, Rongjing Shen 1, Mengting Qian 1, Zhengjun Zhou 2, Bingqing Xie 3, Yong Jiang 3, Yang Yu 1,4,* , Wei Dong 1,*
, Wei Dong 1,*1 Key Laboratory of Medical Electrophysiology, Ministry of Education & Medical Electrophysiological Key Laboratory of Sichuan Province, Institute of Cardiovascular Research, Southwest Medical University, 646000 Luzhou, Sichuan, China
2 Department of Neurosurgery, The Affiliated Hospital of Southwest Medical University, 646000 Luzhou, Sichuan, China
3 Laboratory of Neurological Diseases and Brain Function, Institute of Epigenetics and Brain Science, The Affiliated Hospital of Southwest Medical University, 646000 Luzhou, Sichuan, China
4 Department of Human Anatomy and Histoembryology, School of Basic Medical Sciences, Southwest Medical University, 646000 Luzhou, Sichuan, China
Abstract
Alzheimer’s disease (AD) is a neurodegenerative disease characterized by the formation of amyloid plaques, neurofibrillary tangles and progressive cognitive decline. Amyloid-beta peptide (Aβ) monoclonal antibody therapeutic clinical trials have nearly failed, raising significant concerns about other etiological hypotheses about AD. Recent evidence suggests that AD patients also exhibit persistent neuronal loss and neuronal death accompanied by brain iron deposition or overload-related oxidative stress. Ferroptosis is a type of cell death that depends on iron, unlike autophagy and apoptosis. Inhibiting neuronal ferroptosis function is effective in improving cognitive impairment in AD. Notably, new research shows that ferroptosis in AD is crucially dependent on glial cell activation. This review examines the relationship between the imbalance of iron metabolism, the regulation of iron homeostasis in glial cells and neuronal death in AD pathology. Finally, the review summarizes some current drug research in AD targeting iron homeostasis, many novel iron-chelating compounds and natural compounds showing potential AD-modifying properties that may provide therapeutic targets for treating AD.
Keywords
- ferroptosis
- Alzheimer’s disease
- glial cells
- neuron
- lipid peroxidation
Iron is an essential metal cofactor that plays a vital role in many physiological functions in the body. Various physiological processes are regulated by iron in the brain, including functions that contribute to myelin formation, neurotransmitter synthesis and antioxidant enzymes [1]. Maintaining homeostasis of iron is crucial for maintaining human health, as both iron deficiency and iron overload can be harmful. It has been demonstrated that iron is essential for the normal function of neurons, oligodendrocytes, astrocytes and microglia in the central nervous system (CNS) [2]. Abnormal iron metabolism can increase blood-brain barrier (BBB) permeability and neuroinflammation, promoting neurodegenerative processes [3]. Ferroptosis is a novel type of iron-dependent cell death caused by iron overload, which results in iron-dependent accumulation of lipid peroxides and the production of reactive oxygen species (ROS) [4]. This may be a novel mechanism for inducing neuronal death in neurodegenerative diseases such as Alzheimer’s disease (AD).
There is increasing evidence that iron overload and homeostasis dysregulation of iron contribute to neurodegeneration in AD and that dysregulation of metal homeostasis is associated with cell activation and death processes [5, 6]. In an earlier study, histochemistry showed a striking accumulation of iron (III), localized with iron (II) cyanide, which was closely related to senile plaques, neurofibrillary tangles, and neuropil threads; the iron associated with this lesion promotes in situ oxidation and catalyze H2O2-dependent oxidation, indicating that iron accumulation may be an important factor in oxidative damage in AD [7]. Patients with moderate cognitive impairment (MCI) and AD have higher levels of brain iron deposition, according to quantification using susceptibility-weighted imaging [8]. Further evidence suggests that disruption of iron homeostasis and elevated ferritin levels in the cerebrospinal fluid (CSF) are linked to the progression from MCI to AD [9]. Additionally, a meta-analysis involving 2174 patients with AD and 2931 healthy controls showed decreased levels of iron in the blood and increased ferritin in the CSF of patients with AD when compared to levels in controls, suggesting that iron and iron-associated proteins are associated with the risk of AD [10]. Notably, whole-exome sequencing identified protein-coding variants associated with brain iron in 29,828 individuals, revealing several insights into the relationship between brain iron accumulation and AD and identifying multiple genes associated with brain iron accumulation levels [11]. These studies demonstrated a strong correlation between the pathophysiology of AD and iron homeostasis disturbance.
Recently, most research on iron-mediated neurodegenerative diseases has focused
on neurons, ignoring the role of non-neuronal cells, in regulating pathological
processes. Indeed, in an environment of iron metabolism disorder AD, the
crosstalk between glial cells and neurons may form positive feedback, astrocyte
reactive hyperplasia and amyloid-beta peptide (A
AD is a neurodegenerative disease that stands out as a prominent health problem
for elderly individuals worldwide. It has become evident that AD is not only
linked to its hallmark lesions—the accumulation of A
Aberrant iron uptake, excretion and storage in neurons increases intracellular labile iron pools (LIP) and Fenton responses during the regulation of iron homeostasis [17]. The primary regulator of intracellular iron metabolism is the iron regulatory protein (IRP) response element (IRE) regulatory system [18]. In an intracellular environment deficient in iron, IRP binds to ferritin and homologous IRE in the untranslated region of transferrin receptor 1 (TfR1) mRNA to promote ferritin mRNA translation inhibition and stabilize TfR1 mRNA to prevent intranuclear catabolic events [19]. However, in iron-rich cells, IRP does not bind to the IRE, enhances ferritin synthesis, permitting degradation of TfR1 mRNA and the extracellular transport of Fe3+ by transferrin (Tf) to cells, subsequently reducing Fe2+ [20, 21]. Fe2+ are then transported from endosomes to the cytoplasm via divalent metal transporter 1 (DMT1), which is regulated by homeostasis of LIP [22]. DMT1 is used for normal neuronal metabolism and is partially stored in ferritin to protect neurons from excess iron [23]. When excess H2O2 in the cell interacts with Fe2+, it starts the Fenton reaction, which causes intracellular lipid peroxidation to produce ROS and ferroptosis [24]. Notably, iron export in neurons is mediated by iron-exporting transmembrane protein ferroportin1 (Fpn1) through the Fpn1/Hephaestin and Fpn1/Ceruloplasmin (CP) pathways [25, 26]. Alterations to this pathway induce iron retention, leading to memory impairment [27, 28]. Simultaneous Fe2+ and Fe3+ imaging shows their enrichment in the brains of mice with AD [29]. Moreover, increased ferritin heavy chain (FTH) and ferritin light chain expression were found in the brains of AD patients, suggesting that LIP is increased in AD, confirming that iron can be used as a biomarker for AD [5].
A
The tau protein is a significant component of neurogenic fibrillary tangles,
mediating iron efflux from cells and excess iron colocalizes with tau in
neurogenic fibrillary tangles [31]. Autopsy study finds colocalization of iron
and tau in neurons carrying NFT associated with progressive neurodegeneration
[42]. Neuronal iron overload causes tau hyperphosphorylation by activating
protein kinases, such as glycogen synthase kinase-3
Dysregulation of iron metabolism in AD has been demonstrated mainly in
preclinical studies. Studies have found that Fpn1 is downregulated in AD patients
and APP/PS1 animal models, while genetic absence of Fpn1 resulted in hippocampus
atrophy and memory impairment. However, administration of specific iron chelating
inducer significantly reduces neuronal death and memory deficits caused by
A
Calcium-mediated oxidative stress is largely inseparable from iron, as both iron and Ca2+ play key roles in neuronal function, such as ROS production, mitochondrial homeostasis, and synaptic plasticity [58]. Iron-induced neuronal ROS production alters Ca2+ signaling, which in turn affects synaptic plasticity and hippocampus-dependent memory formation [59, 60]. In the mouse hippocampal neuronal cell line HT-22, excess iron leads to mitochondrial fragmentation and increased Ca2+ levels, which in turn stimulates the Ca2+-dependent phosphatase calcineurin and neuronal cell death [61]. In primary hippocampal neurons, iron-induced ROS increase promotes the release of calcium in the endoplasmic reticulum (ER) and subsequent mitochondrial fission may lead to neuronal dysfunction [62]. Thus, mitochondria as a potential target for the treatment of neurodegenerative processes induced by abnormal iron metabolism. Notably, the crosstalk between iron, ROS, and Ca2+ signaling is bidirectional, as many proteins involved in cellular antioxidant defense and ROS production are dependent on Ca2+. The aberrant increase in intracellular Ca2+ promotes the uptake of mitochondrial Ca2+ by mitochondrial Ca2+ unidirectional transporters, resulting in more ROS production, which may further increase an unstable iron pool [63]. A primary hippocampal neuronal study has reported an influx of iron through Ca2+ channels, resulting in altered intracellular iron content [64]. A sustained increase in Ca2+ flux regulates the immunological profile of ferroptosis [65]. Overall, current evidence suggests that the coordinated interrelationship between excess iron and abnormal Ca2+ signaling is most likely to occur in neurodegeneration, particularly AD, as abnormal calcium signaling is a central issue in AD pathology [66, 67, 68].
Several pathological features found in AD patients are highly correlated with
ferroptosis, including elevated levels of free iron [8], lipid peroxidation [69]
and defects in antioxidant systems such as glutathione peroxidase 4
(GPX4), glutathione (GSH), and cystine/glutamate antiporter system
(system Xc–) [70]. System Xc–, composed of the light chain Solute
Carrier Family 7 Member 11 (SLC7A11) and heavy chain Solute Carrier
Family 3 Member 2 (SLC3A2), is an important glutamate transporter in the
CNS for the cellular uptake of cystine in exchange for intracellular glutamate
[71]. In the context of AD, compensatory uptake of cystine leads to poor
glutamate clearance in the synaptic cleft, causes excitatory neurotoxicity
through system Xc–, enhances lipid peroxidation and leads to ferroptosis and
the development of AD [72, 73]. Additionally, GSH is an important intracellular
antioxidant that prevents iron-dependent oxidation, which is a crucial biological
process in ferroptosis, primarily by binding to Fe2+ in unstable LIP [74].
Reduced GSH levels have been found in the occurrence and progression of AD [75].
Further,
Apolipoprotein E (APOE) allele variants are the most significant
genetic risk for disseminated AD and APOE variants, leading to
A
Ferroptosis in AD is accompanied by multiple pathologic changes. A variety of
other manifestations associated with ferroptosis have been identified in various
neurological diseases mediated by neuroinflammation, and damage-associated
molecular patterns. These include the ROS generated during ferroptosis events
that activate glial cells by activating neuroimmune pathways, which subsequently
lead to the production of various inflammatory cytokines that promote the
development of neurological diseases [91]. Novel drugs that target inflammation
have shown effective intervention against ferroptosis. One study found that
dimethyl fumarate attenuates neuroinflammation and ferroptosis by mediating the
inflammatory central node nuclear factor-
Various pathways, including iron metabolism, redox status, lipid and sugar metabolism, the metabolism of cytotoxic amino acids and mitochondrial activity may regulate the process of ferroptosis [4]. Major markers of ferroptosis, dysregulation of ferritosis, lipid peroxidation and ROS production have long been identified in the brain and animal models of AD patients [96]. A recent transcriptomic study has identified differential expression of genes associated with ferroptosis in AD in different glial cells and neurons [97], suggesting that the pathogenic process of ferroptosis in AD may include glial cells. Though it’s yet unknown exactly how ferroptosis occurs in neurons, non-neuronal cells such as glia are integral to mediating ferroptosis to neurons [17]. When activated, glial cells can induce ferroptosis by disrupting iron homeostasis, releasing proinflammatory factors, altering amino acid metabolism and raising levels of oxidative stress, which are associated with many organ injuries and degenerative diseases [98, 99]. For instance, neurofilament light chain, a pathologic marker for various neurological disorders, including AD, is part of the axonal cytoskeletal protein complex and FTH may be released into the extracellular space while reflecting the senescent state of microglia. After acting on microglia with various neurofilament light chain concentrations, the secretion of FTH-containing exosomes by microglia increased and stimulated peroxidation of membrane lipid and neuronal loss involved in neuronal ferroptosis [100]. Although iron homeostasis and ferroptosis are crucial in causing AD neuronal death, how they interact and lead to neurodegeneration remains poorly understood. Therefore, the pathological processes of iron metabolism disruption and associated cell death urgently require further investigation in the brain.
Microvascular endothelial cells (BMECs), astrocytes, microglia and pericytes
together constitute the BBB [101]. The BBB is an effective protective filter for
the brain as it regulates iron entry, separating cerebral iron homeostasis from
whole-body homeostasis. Expressed on the luminal side of cerebral capillaries,
TfR1 mediates iron uptake in the brain and binds circulating transferrin to
promote the transfer of iron to BMECs through the TfR1-mediated endocytosis
mechanism [3]. Notably, hepcidin is a peptide hormone implicated in the
regulation of Fpn1 and brain iron metabolism. It has been shown that hepcidin is
expressed in cortical neurons, glial cells and BMECs and plays an important role
in the regulation of iron homeostasis [102]. Hepcidin binds to Fpn1 in the cell
membrane of BMECs, promotes the internalization and degradation of Fpn1 and
blocks the passage of iron into the brain through the BBB [103]. It was shown
that hepcidin expression levels were reduced in AD patients and APP transgenic
mice, whereas hepcidin treatment of cultured BMECs and neurons resulted in a
reduction of iron input (TfR1 and DMT1) and output (Fpn1) proteins, which reduced
iron uptake in neurons [104]. Furthermore, in a recent study of the APP/PS1 mouse
model, it has been shown that hepcidin overexpression in astrocytes induces a
reduction in iron levels in cortical and hippocampal neurons, with significant
improvements in cognition and A
Glial cells are not only the primary support cells for neurons but are active
participants in neuronal network formation and information processing in the CNS
[107]. Extensive research has demonstrated that the crucial role of ferroptosis
in the inflammatory response. Ferritin synthesis is regulated by proinflammatory
cytokines, including interleukin-1
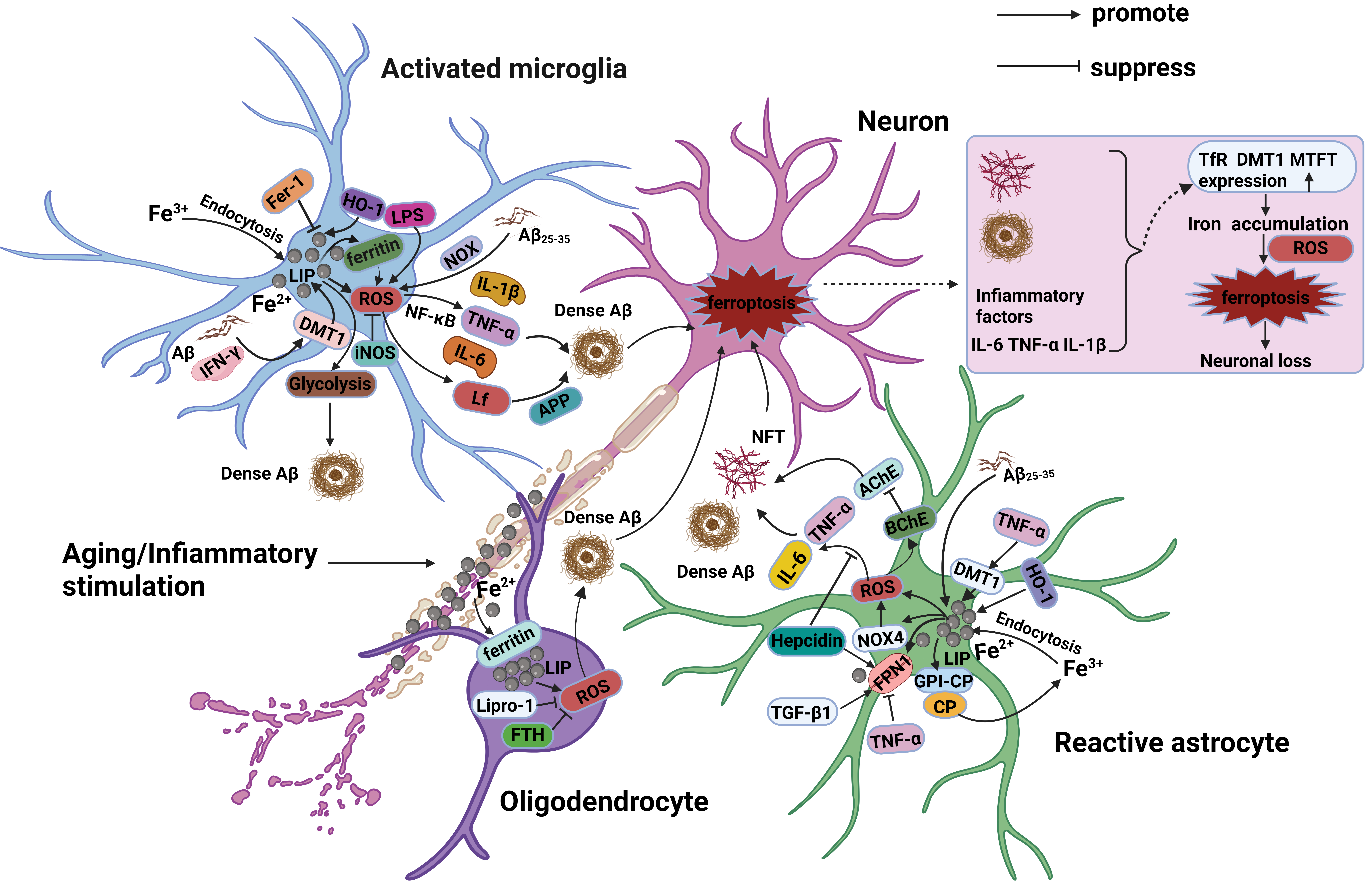 Fig. 1.
Fig. 1.
Molecular mechanisms of glial cell ferroptosis regulation in
AD. In an AD pathological setting, glial cells are exposed to elevated levels of
iron, lipopolysaccharide and extracellular A
As highly reactive immune cells in the brain, microglia play a critical role in regulating the iron metabolism in brain networks [121]. Proteins that regulate iron metabolism in microglia, including transferrin receptors (TfR), ferritin, divalent metal DMT1 and Fpn1, which mediate iron influx from BMECs and the brain interstitium control the amount of iron in neurons [122, 123]. After being released from Tf, iron is translocated to the cytoplasm via DMT1 or other transporters [124]. Thus, microglia can internalize non-Tf-bound iron (NTBI) and Tf iron through different uptake pathways [125]. Microglia regulate their iron levels through the IRP and IRE signaling pathways, which control the expression of proteins related to iron metabolism; when intracellular iron levels are low, IRP binds to transferrin receptors and iron IRE in ferritin mRNA, leading to an increase in the expression of these proteins and promoting iron uptake and storage [125]. Under conditions of neuroimmune activation, the abundance of DMT1 on the surface of microglia and neurons is increased, and these cells are susceptible to ferroptosis due to iron overload induced by high levels of NTBI [126].
First, the microglia of activated cells can accumulate iron, leading to
increased iron accumulation in the CNS within the brain [127]. The study reported
that iron supplementation in newborn mice promotes neurodegeneration, leading to
microglia activation, increased volume and reduced process length [128]. Second,
proinflammatory cytokines mediated by NF-
In the APP/PS1 mouse, an inflammatory microglia phenotype was induced with
IFN-
Astrocytes, a component of the BBB, are thought to be critical regulators of
redox-active metal-iron metabolism within the brain and they influence iron
homeostasis by regulating the transfer of iron from the outer body into the CNS,
as well as the inflow and outflow of iron between neurons and glial cells [146].
Since the terminal foot protrusions of astrocytes are closely linked to the
luminal creation of BMECs, this may be the primary pathway for iron to enter
brain tissue via the Tf/TfR1 pathway [147]. Studies show astrocytes exhibit high
levels of DMT1 and TfR1, as well as the iron oxidase CP, iron inflow and iron
efflux proteins [148, 149]. Among them, DMT1 is predominantly expressed in
astrocytes associated with vascular endothelial cells, which play a major role in
iron uptake [149]. Moreover, CP acts as an iron oxidase, oxidizes Fe2+ to
Fe3+, promotes iron efflux from cells to regulate iron homeostasis, inhibits
lipid peroxidation mediated by the instability of the pool of intracellular
Fe2+ and prevents neurotoxicity [150]; in the absence of physiological
activity of CP proteins, Fe2+ cannot be oxidized, leading to a significant
iron accumulation in astrocytes and loss of glial-derived growth factors
essential for neurons [151]. CP knockout mice lead to progressive astrocyte and
neuronal death and contribute to neurodegenerative processes [152]. According to
recent research, the CSF of AD patients contains substantially less CP than
normal [153]. In A
Unlike CP, heme oxygenase-1 (HO-1) breaks down heme into carbon monoxide, free
iron and biliverdin [157]. It acts as a sensor of mitochondrial iron chelation in
astrocytes, and overexpression of HO-1 in astrocytes contributes to the
pathological deposition of non-transferrin iron and adjacent neuronal damage. In
contrast, using the iron chelator DFO effectively reduces the death of cultured
astrocytes [158]. Furthermore, it was discovered that, in comparison to controls,
HO-1 was substantially more immunoreactive in astrocytes and neurons in the
cerebral cortex and hippocampus and it colocalized with neurogenic fibrous
tangles and amyloid senile plaques in AD patients [159]. This suggests neuronal
death in AD pathogenesis is related to elevated astrocyte expression of HO-1 and
consequent iron accumulation. Within the brain’s glia and white matter,
butyrylcholinesterase (BChE) participates in cholinergic neurotransmission
alongside acetylcholinesterase [160]. In AD mice, pathogenic A
Further, given that astrocytes are Cu depots, dysregulation of Cu metabolism in the brain is characteristic of neurodegenerative diseases, including AD [164]. The cytotoxicity of copper disrupts mitochondrial integrity by aggregating lipoylated proteins and causing the loss of Fe-S cluster proteins, thereby triggering cuproptosis, leads to oxidative stress and mitochondrial dysfunction, which has something in common with the pathological mechanism of ferroptosis [165]. In fact, in addition to the mitochondrial tricarboxylic acid cycle, GSH is a common hub of ferroptosis and cuproptosis, albeit with different roles, in ferroptosis GSH acts as an antioxidant, preventing lipid peroxidation, while in cuproptosis it acts as a copper companion, binding copper to mitigate the aggregation of lipoylated proteins [166]. The depletion of GSH levels is a feature of brain aging and has been associated with the progression of neurodegenerative diseases, including impaired cognitive function and ferroptosis in AD patients [167]. Therefore, it may be speculated that cuproptosis and ferroptosis may have a cross talk in AD, but the upstream and downstream molecules that mediate the common pathological mechanism need to be further studied.
The brain’s oligodendrocytes are the cells with the highest iron content. Iron
is bound by oligodendrocytes as metabolic substrates for myelin synthesis and
maintenance; the formation of myelin sheaths is correlated with both direct and
indirect iron uptake [168]. Indeed, ferritin is found in AD patients, mainly in
oligodendrocytes and myelinated axons of glial dystrophy [169]. By comparing
cortical myelin formation profiles with A
Molecular mechanisms of ferroptosis in AD have been implicated in increased
oxidative stress. Neuronal networks are attenuated by glial cells via a variety
of pathways that inhibit the entry of excess ROS into neurons, with the targets
they provide being valuable for restoring the degenerating metabolic status of
neurons. Activation of microglia induces neuronal ferroptosis in AD through
increased oxidative stress. One study found that A
When reactive, astrocytes release a variety of antioxidant molecules such as
GSH, hepcidin and Nrf2, which are critical regulators in iron metabolism and
oxidative stress neuroprotection [112, 179, 180]. Hepcidin is one of the key
regulators of systemic iron homeostasis; astrocyte hepcidin regulates iron uptake
at the BBB and hepcidin injection into the brain attenuates iron deposition
[105]. In an A
Oligodendrocyte death, dysfunction and/or demyelination occur in AD [185]. It has been estimated that oligodendrocytes operate at the highest metabolic rate among brain cells that consume a lot of oxygen to generate adenosine triphosphate to provide energy for the production of myelin sheaths, resulting in the production of hydrogen peroxide and other ROS [186, 187]. Iron serves as an important cofactor in myelin synthesis; it provides raw material for myelin production and tends to trigger the Fenton reaction, leading to enhanced lipid peroxidation and promotion of ferroptosis [188]. Paradoxically, low antioxidant content in oligodendrocytes such as GSH further amplifies ferroptosis. A study has shown that liproxstatin-1 prevents ferroptosis; the use of liproxstatin-1 not only restored the expression of GPX4, GSH and iron death inhibitory proteins, but also inhibited oligodendrocyte mitochondrial lipid peroxidation and GPX4-induced iron death in oligodendrocytes [189]. This suggests that antioxidants targeting ferroptosis with age may be the direction of research to target non-neuronal cells. Since iron accumulation is associated with many neurological diseases, exploring oligodendrocytes and AD pathogenesis in future studies should be encouraged.
One key mechanism causing AD neurodegeneration is ferroptosis. Therefore, in
patients with AD, targeting ferroptosis may be a promising treatment option.
Inhibitors of ferroptosis with current applications for treating and preventing
AD are emerging (Table 1, Ref. [73, 105, 190, 191, 192, 193, 194, 195, 196, 197, 198, 199, 200, 201, 202, 203, 204, 205, 206, 207, 208, 209, 210, 211, 212, 213, 214]). Therapeutic effects in AD
models have been demonstrated by iron chelators that are used in clinical
settings, such as DFO, deferiprone (DFP) and deferasirox [215, 216]. In a
24-month, a single-blind trial of 48 patients with AD, intramuscular injection of
DFO was found to reduce the rate of cognitive decline in these patients by 50%
when compared to the normal controls [190]. DFO is challenging to transport
across the BBB, has low bioavailability, the nasally administered DFO
nanoparticles under investigation facilitate brain transport [217] and have a
potential role in AD treatment. DFP readily crosses the BBB and chelates
intracellular iron. It is currently being studied in a phase II randomized,
double-blind, placebo-controlled clinical trial (ClinicalTrials.gov ID
NCT03234686) designed to investigate the safety and efficacy of DFP in subjects
with MCI, prodromal AD and mild AD [218]. DFP has a high safety profile, low systemic
toxicity and is a viable strategy for anti-ferroptosis therapy in AD.
Additionally, other iron chelators, such as chloroioquine and its derivatives,
may also help with the treatment of AD by reducing tau and A
| Sort | Drug candidates | Key results | References |
| Iron Chelators | Deferoxamine | Cognitive decline in AD patients slowed by 50% | [190] |
| Deferiprone | Phase AD II clinical trials are ongoing; Reverses pathological brain iron accumulation; Reduces iron deposition, A |
[191, 192] | |
| Deferasirox | Blocks iron accumulation, reduces expression of ferritin and transferrin receptors; reverses changes in A |
[193, 194] | |
| Clioquinol | Prevention of cognitive decline in 32 AD patients and reduction of plasma A |
[195, 196] | |
| M30 | Reduces APP expression and A |
[197, 198] | |
| HPO | Neuroprotective effect on A |
[199] | |
| Hydroxylated chalcones | Fights human neuroblastoma SH-SY5Y cells A |
[200] | |
| HLA20A | Reduces APP expression and A |
[201] | |
| Tacrine | Neuroprotective in neuroblastoma cells treatment with A |
[202, 203] | |
| Schiff bases | Inhibit redox active metals and metal-induced A |
[204] | |
| Antioxidants | Selenium | Inhibit ferroptosis. Key regulator of the activity of GPX4. Improvement of cognitive impairment in AD animal models | [205, 206] |
| Vitamin E | Lipophilic free radicals capture antioxidants, fight ferroptosis, improve lipid ROS to prevent iron denaturation in GPX4-deficient cells, slows cognitive deterioration in patients with AD | [207] | |
| Alpha-Lipoic Acid | Blocks tau induced iron overload and lipid peroxidation and improves cognitive function in AD patients. Slowed cognitive decline in 129 probable AD patients | [208, 209] | |
| Ferrostatin-1 | Prevents neuronal death and memory loss. Reduces iron deposition and neuronal degeneration. Improves long-term motor and cognitive function | [73, 210] | |
| Liproxstatin-1 | A |
[73] | |
| Hepcidin | Reduces iron and A |
[105] | |
| Polyphenols | Reduces iron accumulation, clears ROS, inhibits A |
[211, 212] | |
| FSP1 | A FSP1-CoQ10-NAD(P)H pathway, along with GPX4 and GSH, inhibits ferroptosis and phospholipid peroxidation | [213] | |
| NQO1 | Inhibits lipid peroxidation and maintains antioxidant forms of |
[214] |
AD, Alzheimer’s disease; CoQ10, Coenzyme Q10; GSH, glutathione; GPX4, glutathione peroxidase 4; NQO1, NAD(P)H Quinone Dehydrogenase 1; FSP1, Ferroptosis suppressor protein-1; HPO, hydroxypyridin-4-ones.
Traditional metal chelators have a single function in terms of treatment and as
previously mentioned that there may be crosstalk in the pathogenesis of AD with
cuproptosis and ferroptosis, dual chelating agents like Cu chelators along with
Fe chelatore, have also been studied in the treatment of AD.
Notably, some genes targeting Fe chelators along with the gene responsible for promoting ferroptosis or anti-ferroptosis genes should be considered. For example, NCOA4, as a selective autophagy receptor, has been identified to colocalize with endogenous LC3B and intracellular iron storage ferritin complexes. However, NCOA4-mediated ferritosis-induced ferroptosis dysfunction can accelerate the pathological process of AD [230], which suggests that targeting NCOA4-mediated ferritinophagy holds promise for preventing and treating AD. Recently, the role of Nrf2 as a key regulator against oxidative stress has attracted much attention. Nrf2 exerts an antiferroptosis role by regulating the expression of a large number of ferroptosis-related genes and key aspects of lipid metabolism, which play an important role in the development of AD, as brain cells are rich in lipid [231]. Future studies should examine the mechanisms of iron homeostasis dysregulation and ferroptosis, as well as the role of the Nrf2-regulated ferroptosis-related signaling pathway in the development of AD, both to determine new drugs that effectively treat AD and identify their key targets.
Despite the compelling evidence supporting iron’s involvement in the pathogenesis of AD, it is unclear what roles iron play both upstream and downstream in the pathophysiology of AD. More recently, a growing number of iron-chelating compounds and antioxidants have been found to show potential therapeutic benefits. Although it is still unclear as to whether these ferroptosis inhibitors are effective in treating AD patients, their performance has demonstrated encouraging results in AD models and future research into the use of drugs with these potential therapeutic properties (e.g., iron chelators and multifunctional metal chelators) and their to entry into clinical trials, improved bioavailability and ability to effectively cross the BBB into the brain for practical physiological functions will be a significant issue in ferroptosis-mediated AD therapeutic pharmacology research.
Given an aging population, early detection of AD is essential. Notwithstanding substantial progress in understanding the etiology of AD, drug development therapeutic strategies based on these discoveries have very limited efficacy in altering the course of the disease. Consequently, there are compelling reasons to investigate new therapeutic approaches. Ferroptosis research has recently made rapid progress in understanding the pathophysiology of AD. Dysregulation of brain iron homeostasis exacerbates neurotoxicity and cognitive impairment. Thus, targeted ferroptosis inhibitors gain new momentum in the treatment of AD. Nevertheless, further study is required to pinpoint the exact cause of neurodegenerative diseases linked to ferroptosis. The current review elucidates the function of iron homeostasis between neuronal and glial cells in AD pathogenesis. Disturbed iron metabolism in glial cells mediates inflammation, oxidative stress and other pathological processes damaging neurons; iron deposition in neurons also leads to glial cell hyperactivation, accompanied by the release of proinflammatory factors and iron metabolism molecules by overactive microglia and astrocytes, mediating neuronal cell death in AD pathology. There are few studies on the mechanism of ferroptosis in oligodendrocytes in AD. Oligodendrocytes are high in iron content and tightly wrapped around neuronal axons; further study is necessary to investigate the ferroptosis induced by oligodendrocytes in AD neurons during aging.
In summary, ferroptosis is involved in the interaction between glial cells and neurons, thereby regulating the pathogenesis of AD. However, experimental animal models are the primary focus of current ferroptosis research and changes in ferroptosis in AD patients cannot be fully simulated. Comprehensive preclinical investigations are necessary to completely understand the precise molecular and cellular pathways of iron metabolism in AD neuronal degeneration, even while clinical trials assess the potential treatment benefits focused on ferroptosis.
YY and WD conceived the perspective of the work. JX, RS, MQ, and BX drafted the manuscript. JX, ZZ, BX, and YJ designed the figure. RS and MQ collect and sort references. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This work was supported by the National Natural Science Foundation of China (31871031, WD), Ministry of Science and Technology of the People’s Republic of China (2019YFE0120600), and Sichuan Science and Technology Program (2023NSFSC0593, YY; 2022YFS0615, BX), the Science and Technology Planning Project of Luzhou (2022-JYJ-140 to YY).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.