


 , Xuebin Wang 2,3,†, Yahui Li 1,4, Jing Zhao 1,5, Xue Zhou 2,6,*
, Xuebin Wang 2,3,†, Yahui Li 1,4, Jing Zhao 1,5, Xue Zhou 2,6,* , Xingchen Wang 3,7,*
, Xingchen Wang 3,7,*1 First Clinical Medical College, Shandong University of Traditional Chinese Medicine, 250014 Jinan, Shandong, China
2 Postdoctoral Research Station, Shandong University of Traditional Chinese Medicine, 250014 Jinan, Shandong, China
3 Department of Neurology, The Second Affiliated Hospital of Shandong University of Traditional Chinese Medicine, 250001 Jinan, Shandong, China
4 Department of Gerontology, The Second Affiliated Hospital of Shandong University of Traditional Chinese Medicine, 250001 Jinan, Shandong, China
5 Experimental Center, Shandong University of Traditional Chinese Medicine, 250399 Jinan, Shandong, China
6 Division of Neurology, Affiliated Hospital of Shandong University of Traditional Chinese Medicine, 250014 Jinan, Shandong, China
7 The Second Clinical Medical College, Shandong University of Traditional Chinese Medicine, 250001 Jinan, Shandong, China
†These authors contributed equally.
Abstract
Ischemic stroke (IS) constitutes a major threat to human health. Vascular recanalization by intravenous thrombolysis and mechanical thrombolysis remain the most significant and effective methods for relief of ischemia. Key elements of these treatments include achieving blood-vessel recanalization, restoring brain-tissue reperfusion, and preserving the ischemic penumbra. However, in achieving the therapeutic goals of vascular recanalization, secondary damage to brain tissue from cerebral ischemia-reperfusion injury (CIRI) must also be addressed. Despite advancements in understanding the pathological processes associated with CIRI, effective interventions to prevent its onset and progression are still lacking. Recent research has indicated that mitophagy and ferroptosis are critical mechanisms in the development of CIRI, and significantly contribute to the onset and progression of IS and CIRI because of common targets and co-occurrence mechanisms. Therefore, exploring and summarizing the potential connections between mitophagy and ferroptosis during CIRI is crucial. In the present review, we mainly focused on the mechanisms of mitochondrial autophagy and ferroptosis, and their interaction, in the development of CIRI. We believe that the data show a strong relationship between mitochondrial autophagy and ferroptosis with interactive regulation. This information may underpin new potential approaches for the prevention and treatment of IS and subsequent CIRI.
Keywords
- cerebral ischemia-reperfusion injury
- mitophagy
- ferroptosis
- ischemic stroke
- mechanism research
Ischemic stroke (IS) results from impaired brain blood supply due to various cerebrovascular conditions; it leads to brain-tissue ischemia, anoxic necrosis, and rapid onset of neurological deficits [1, 2, 3]. Epidemiological data [4] reveal that deaths attributed to IS increased from 2.04 million to 3.29 million globally, between 1990 and 2019, with projections indicating a rise to 4.9 million by 2030. As the second leading cause of death worldwide, IS is associated with high mortality and disability rates, significantly burdening health systems and economies [5, 6].
The primary pathogenesis of IS involves stenosis or occlusion of intracranial blood vessels caused by various risk factors, leading to either temporary or permanent interruption of cerebral blood flow. This disruption in circulation results in ischemic and hypoxic damage to neurons, culminating in necrosis [3, 7]. Consequently, the cornerstone of IS treatment is achieving vascular recanalization, restoring reperfusion to brain tissue, and preserving the ischemic penumbra [8]. It is important to note that one study [9] has indicated that blood-system diseases may also contribute to the occurrence of IS. Although this scenario accounts for less than 1% of all strokes, it should be acknowledged and considered in the clinical diagnosis and treatment process. Upon identifying the underlying cause of the condition, a suitable treatment plan can be developed.
In recent years, advancements in vascular-reperfusion strategies have led to the continuous refinement of therapeutic drugs and surgical techniques, thereby increasing the proportion of patients benefiting from these treatments [10]. However, reperfusion after brain-tissue ischemia often induces secondary damage, known as cerebral ischemia-reperfusion injury (CIRI) [11]. Even with timely reperfusion therapy, patients may still experience severe neurological deficits. CIRI is a key pathophysiological process in neurological diseases like IS, involving complex mechanisms such as oxidative stress [12], inflammatory responses [13, 14], Ca2+ overload [15, 16], nitric oxide injury [17, 18, 19, 20, 21], excitotoxicity [22, 23], mitophagy [11], and ferroptosis [24, 25]. Among these, mitophagy and ferroptosis are particularly significant in CIRI and have become prominent areas of research in recent years.
Mitophagy is the selective removal of damaged mitochondria, thus crucially maintaining cellular homeostasis and preventing disease onset [26]. During CIRI, mitophagy primarily acts neuroprotectively by eliminating damaged mitochondria and modulating inflammatory responses [27]. Ferroptosis is a form of cell death driven by iron-dependent lipid peroxidation. In CIRI, iron accumulation in brain tissue signals this process [28], leading to oxidative stress through abnormal iron metabolism, dysregulated amino acid metabolism, and lipid peroxidation. This cascade ultimately disrupts cell and organelle membranes, triggering neuronal apoptosis or necrosis. Both mitophagy and ferroptosis are integral to CIRI pathology and are closely interrelated. Mitochondria are common targets for both processes, with oxidative stress as their shared mechanism. These processes are interconnected by various “bridges”, and exert mutual regulatory effects [29]. Understanding the interaction between mitophagy and ferroptosis in CIRI is essential. This review aims to elucidate the mechanisms of mitophagy and ferroptosis, and their interactive dynamics, involved in CIRI, offering a novel perspective on treating IS and mitigating CIRI.
We searched the Web of Science and PubMed databases up to August 2024 for original research articles related to the mechanisms of mitophagy and ferroptosis in the occurrence of CIRI. The keywords used were ‘cerebral ischemia-reperfusion injury’, ‘mitophagy’, ‘ferroptosis’, ‘mechanism’, and ‘oxidative stress’.
Mitochondria, the energy centers of all eukaryotic cells, are essential for adenosine triphosphate (ATP) production via aerobic respiration and oxidative phosphorylation [30]. Their function is essential for cellular homeostasis [31]. They are closely linked to mitochondrial membrane potential [32], mitochondrial Ca2+ concentration [33], respiratory chain complex activity [34], reactive oxygen species (ROS) generation [35], and mitochondrial DNA mutations [36]. Mitochondrial quality control ensures mitochondrial integrity and functionality through coordinated regulation of protein balance [37], mitophagy [38], mitochondrial dynamics [39], and biogenesis [40]. Excessive ROS accumulation causes mitochondrial damage, thereby impairing mitochondrial DNA, altering membrane potential, and modifying oxidized protein activity, ultimately disrupting cellular metabolic homeostasis and leading to cell death. To maintain cellular and mitochondrial homeostasis and prevent damage, cells selectively encapsulate and degrade dysfunctional mitochondria through mitochondrial autophagy (mitophagy).
Mitophagy is a critical mitochondrial quality-control mechanism [41], significantly regulating structural and functional mitochondrial damage in neural cells due to CIRI. This process involves four key steps [42]: (1) mitochondrial damage triggers permeability transition, leading to depolarization, loss of membrane potential, and activation of mitophagy-related proteins; (2) damaged mitochondria are enveloped by autophagosomes, forming mitophagosomes; (3) mitophagosomes fuse with lysosomes to create mature mitophagy-lysosomes; and (4) lysosomal or vacuolar acid hydrolases degrade the contents of the mitochondria within autophagosomes, ensuring the removal of damaged mitochondria.
The mitophagy mechanism involves two primary pathways: the ubiquitin (Ub)-dependent and the Ub-independent pathways [41]. Central to the Ub-dependent pathway are the proteins phosphatase and tensin homolog-induced kinase 1 (PINK1) and Parkinson protein 2 E3 ubiquitin-protein ligase (Parkin) [43]. The PINK1/Parkin system is pivotal in eliminating damaged mitochondria [44]. Under normal conditions, newly synthesized cytoplasmic PINK1 is imported into mitochondria via the mitochondrial targeting sequence. It is cleaved by mitochondrial matrix processing peptidase (MPP) and mitochondrial presenilin-associated rhomboid-like protein (PARL), then released into the cytoplasm for proteasomal degradation. However, when mitochondria are damaged, the decreased inner mitochondrial membrane potential prevents PINK1 importation, leading to its accumulation on the outer mitochondrial membrane [45]. PINK1 then undergoes autophosphorylation and activation, subsequently phosphorylating Ub on the outer membrane, which recruits and activates Parkin [46]. This activation induces a conformational change in Parkin, transforming it into an active E3 Ub ligase [47]. Parkin ubiquitinates mitochondrial outer membrane proteins, and PINK1 phosphorylates the Ub chains, forming phosphorylated Ub-modified proteins. These modified proteins act as “eat me” signals, recognized by autophagy adapter proteins such as sequestosome-1 (p62), nuclear dot protein 52 (NDP52), and optineurin (OPTN). These adapters bind to microtubule-associated protein 1 light chain 3 (LC3), facilitating the encapsulation of damaged mitochondria into autophagosomes, forming mitophagosomes. The mitophagosomes then fuse with lysosomes, in which lysosomal acid hydrolases degrade the mitochondrial contents, effectively clearing the damaged mitochondria. Furthermore, OPTN and NDP52 can function independently of Parkin, with PINK1 directly recruiting them via Ub phosphorylation, thus aiding autophagy biogenesis [48]. In the Ub-independent pathway, protein receptor-mediated mitophagy involves specific outer mitochondrial membrane receptors such as NIP3-like protein X/B-cell lymphoma-2 (BCL2)/adenovirus E1B 19kDa protein interacting protein 3-like (NIX/BNIP3L) [49, 50], BCL2/adenovirus E1B 19kDa interacting protein 3 (BNIP3) [50], and FUN14 domain containing protein 1 (FUNDC1) [51], which directly bind to LC3, inducing mitophagy. Under conditions like hypoxia, these receptors are transcriptionally activated, forming homodimers anchored to the outer mitochondrial membrane. They directly bind to LC3, recruiting damaged mitochondria to autophagosome formation sites. This process promotes autophagosome encapsulation and formation, thereby completing mitophagy. Collectively, these pathways synergize to maintain mitochondrial and cellular homeostasis (Fig. 1).
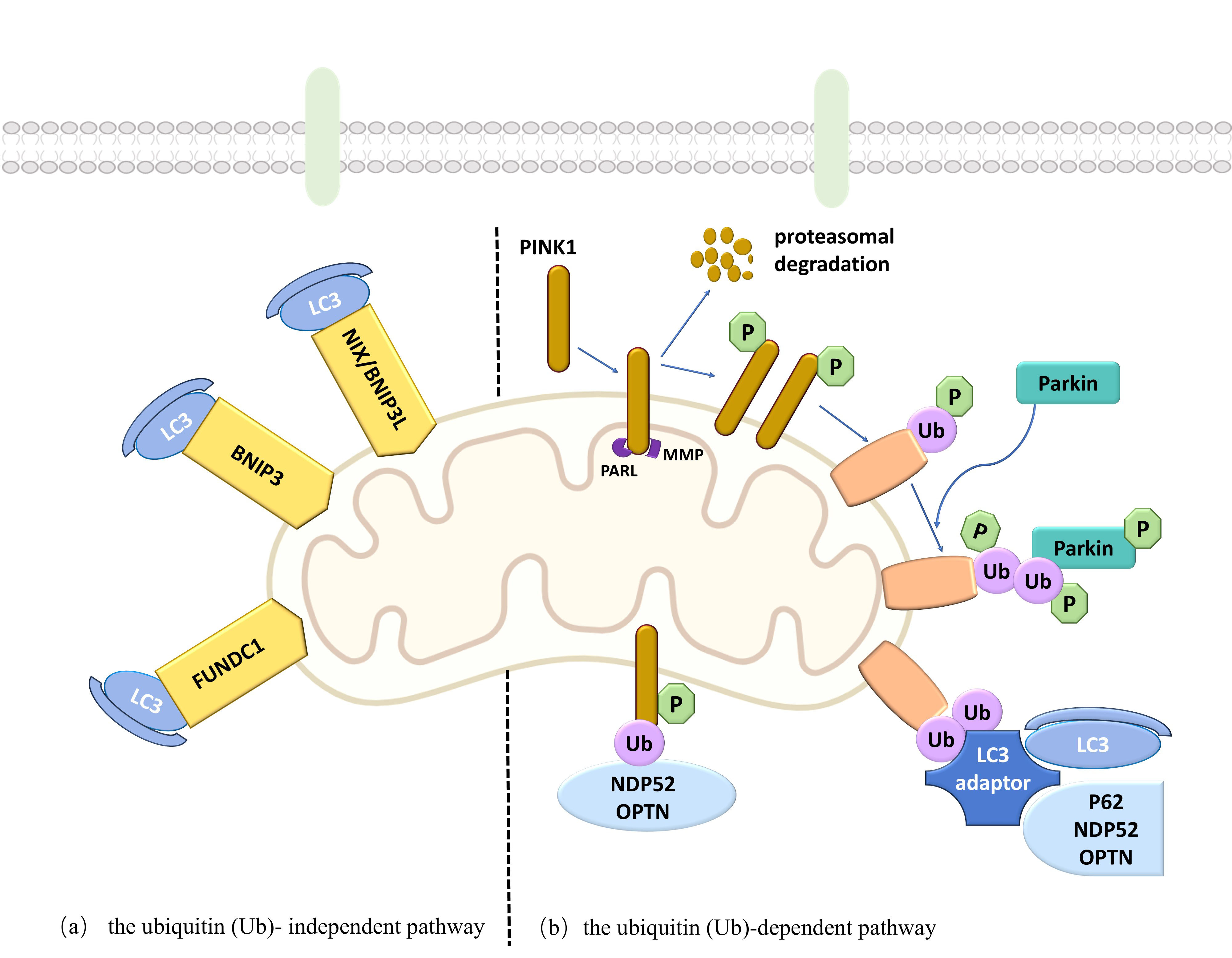 Fig. 1.
Fig. 1.
Mechanism diagram of mitophagy. The mitophagy mechanism encompasses two primary pathways: the ubiquitin Ub-dependent and the Ub-independent pathways. BNIP3, B-cell lymphoma-2 (BCL2)/Adenovirus E1B 19kDa interacting protein 3; FUNDC1, FUN14 domain containing protein 1; LC3, microtubule-associated protein 1 light chain 3; MMP, mitochondrial membrane potential; NDP52, nuclear dot protein 52; NIX/BNIP3L, NIP3-like protein X/BCL2/adenovirus E1B 19kDa protein interacting protein 3-like; OPTN, Optineurin; p62, sequestosome-1; Parkin, Parkinson protein 2 E3 ubiquitin-protein ligase; PARL, presenilins-associated rhomboid-like protein; PINK1, phosphatase and tensin homolog-induced kinase 1. Created the figure with Microsoft Office PowerPoint.
ROS comprise various oxygen-containing reactive compounds [52], primarily generated at the substrate end of the mitochondrial respiratory chain within the inner membrane [35]. Essential for maintaining the dynamic balance between oxidative and antioxidant systems, ROS are regulated through both enzymatic and non-enzymatic mechanisms. Although critical for cell proliferation, hypoxic adaptation, and cell fate determination [53], excessive ROS can cause cell damage and cell death [54]. In ischemia, insufficient oxygen and glucose transport inhibits mitochondrial respiration, leading to irreversible damage and dysfunction, which disrupts ROS homeostasis [55]. Upon reperfusion, the sudden influx of oxygen generates significant oxidative stress products, causing rapid accumulation of free radicals, which damages endothelial cells and capillary pericytes. This damage results in cerebral microcirculation disorders and blood brain barrier (BBB) disruption. The interplay between rising ROS levels and mitochondrial damage triggers “ROS-induced ROS production”, further exacerbating cellular injury [56].
Mitochondria are the primary sites for cellular energy production and Ca2+ buffering, essential for oxidative phosphorylation and ATP synthesis. Reduced ATP disrupts intracellular ion homeostasis. After IS, glucose and oxygen necessary for normal cellular metabolism decrease, leading to decreased ATP levels in the brain, causing dysfunction of cell membrane ion transporters and increased membrane permeability. Consequently, extracellular Ca2+ enters the cell along its concentration gradient. Restoration of blood flow to ischemic tissue elevates intracellular ROS, Ca2+ overload, and production of pro-inflammatory factors and active enzymes, triggering brain cell apoptosis. CIRI destabilizes mitochondrial membrane potential (MMP) and redox transitions, adversely affecting mitochondrial integrity and cell survival. MMP, intricately linked to mitochondrial function, relies on the proper function of the electron transport chain, and its maintenance within a specific range is essential for normal mitochondrial activity [57]. As a sensitive indicator of cellular damage, MMP represents a potential therapeutic target for cerebrovascular diseases. Modulating MMP can reduce neuronal apoptosis, inhibit oxidative stress, and enhance mitochondrial function. Excessive ROS and Ca2+, generated during CIRI, can open the mitochondrial permeability-transition pore (mPTP), altering mitochondrial permeability. This change disrupts ion gradients across the inner membrane, leading to brain cell necrosis and apoptosis [58]. Research has indicated that enhancing mitochondrial function significantly elevates intracellular ATP levels and mitigates damage from hypoxia-reoxygenation injury, improving outcomes for patients with CIRI.
After IS, vascular occlusion disrupts the timely delivery of oxygen and essential energy substrates needed for normal neuron function. Consequently, the cells cannot produce sufficient ATP through standard metabolic pathways; this leads to metabolic disorders. The primary pathological damage produced by cerebral ischemia at this stage includes mitochondrial structural damage and enzyme dysfunction [59]. Although restoring blood flow post-CIRI provides fresh oxygen, it also generates substantial amounts of ROS, which directly harms mitochondria, increases oxidative stress, and causes Ca2+ overload within mitochondria. This cascade triggers the opening of the mPTP and the loss of membrane potential, thereby activating stress-induced mitophagy. Mitophagy removes functionally impaired mitochondria, prevents excessive ROS production and cellular apoptosis, and reduces brain ischemia-reperfusion injury. A recent study has shown that astrocytes can transfer healthy mitochondria to neurons [60]; this process enhances mitochondrial function, minimizes nerve cell and vascular microcirculation damage, promotes neuronal survival, and provides resistance against CIRI.
The inflammatory response significantly contributes to neuron damage in CIRI. Mitophagy plays a protective role by modulating inflammatory signaling pathways and mitigating inflammatory responses. Research has demonstrated [61] that tissue plasminogen activator exerts a neuroprotective effect during CIRI by activating adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK) phosphorylation, enhancing FUNDC1 expression, inhibiting apoptosis, and improving mitochondrial function. Conversely, FUNDC1 overexpression inhibits NOD-like receptor thermal protein domain associated protein 3 (NLRP3) inflammasome activation, reducing inflammation post-brain injury. The NLRP3 inflammasome is a multi-protein complex in the cytoplasm. Mitochondrial dysfunction from CIRI leads to mitochondrial ROS production and mitochondrial DNA (mtDNA) release, activating the NLRP3 inflammasome [62] and triggering an inflammatory response that exacerbates ischemic brain tissue damage. One study has indicated that enhancing mitophagy during CIRI can regulate NLRP3 inflammasome activity, inhibit pro-inflammatory factor release by microglia, and effectively reduce the inflammatory response in nerve cells post-IS, thereby protecting against CIRI injury [63]. That study also confirmed that mitophagy activity is negatively correlated with the intensity of the inflammatory response. Therefore, activating mitophagy can diminish the inflammatory response in IS, thereby improving neuronal damage in ischemic brain tissue.
Mitochondrial dysfunction is intrinsically linked to the vicious cycle of CIRI [64]. After CIRI, Drp1, a key protein regulating mitochondrial fission, becomes activated, leading to increased mitochondrial division and ROS accumulation. This process activates the receptor-interacting protein 1/receptor-interacting protein 3 (RIP1/RIP3) pathway and impedes autophagosome degradation. The resulting undegraded autophagosomes are secreted as exosomes, initiating an inflammatory cascade that further damages mitochondria. This damage leads to excessive ROS generation, further obstructing autophagosome degradation and perpetuating the cycle.
Mitophagy exhibits dual characteristics: moderate mitophagy during CIRI effectively eliminates damaged mitochondria, preserves essential mitochondrial functions, and supports neuronal cell survival [61, 65]. However, excessive mitophagy, while clearing damaged mitochondria, can harm neuronal cells and exacerbate CIRI [66, 67, 68]. Given the dual roles of mitophagy at various pathological stages of CIRI, further investigation is essential to elucidate the specific mechanisms involved. Understanding the induction rate and duration of autophagy activation, when mitophagy exerts beneficial effects, is essential. The role of mitophagy in CIRI is summarized in Table 1 (Ref. [69, 70, 71, 72, 73, 74, 75, 76, 77, 78]).
| Cells or animals | Model | Target | Mechanisms | Effect | Reference |
| Human cerebral microvascular endothelial hCMEC/D3 cells and male C57BL-6/J mice | OGD/R model and MCAO/R model | Upregulate the antioxidant heme oxygenase-1 | 1. Increase mitochondrial ROS formation | Promotion of mitophagy mitigates CIRI | [69] |
| 2. Angiogenesis | |||||
| 3. Reduced cell proliferation | |||||
| 4. Reduced mitochondrial energy metabolism | |||||
| 5. Reduced endoplasmic reticulum stress | |||||
| 6. Increased antioxidant responses | |||||
| HT22 cells | OGD/R model | Upregulate the protein level of PINK1, Parkin, LC3 and Beclin1 and to accelerate the degradation of p62 | 1. Improve mitochondrial dysfunction | Promotion of mitophagy mitigates CIRI | [70] |
| 2. Inhibit apoptosis | |||||
| PC12 cells and SD male rats | OGD/R model and tMCAO/R model | Increased the expression of promoting-mitophagy targets (PINK1, PRKN and LC3B) | Inhibit oxidation reaction | Promotion of mitophagy mitigates CIRI | [71] |
| SH-SY5Y cells and healthy male C57BL/6 mice | OGD/R model and MCAO/R model | Inhibit CK2 |
Reduced oxidative stress and mitochondrial damage | Promotion of mitophagy mitigates CIRI | [72] |
| SD rat brain microvascular endothelial cells | OGD/R model | Upregulate the expression of TRPV4 | 1. Elevate mitochondrial membrane potential | Promotion of mitophagy mitigates CIRI | [73] |
| 2. Increase Table 1 ATP content | |||||
| 3. Improve cell viability | |||||
| Primary neurons were isolated from the brain cortex of embryonic rats and male SD rats | OGD/R model and tMCAO/R model | Upregulate GluN2B, activate downstream CaMKII and ERK1/2 | 1. Inhibit microglia-mediated neuroinflammation | Promotion of mitophagy mitigates CIRI | [74] |
| 2. Regulate mitochondrial function | |||||
| 3. Increase microvascular density | |||||
| 4. Decrease oxidative stress | |||||
| Primary hippocampal neurons isolated from newborn rats and male SD rats | OGD/R model and MCAO/R model | Activate transcription of BNIP3 to enhance BNIP3-mediated mitophagy | 1. Improve the cell survival and prosurvival autophagy | Promotion of mitophagy mitigates CIRI | [75] |
| 2. Reduced cell apoptosis | |||||
| Mouse cerebral microvascular endothelial cells and male SD rats | OGD/R model and MCAO/R model | Elevate of SIRT3 expression | 1. Alleviate neuron apoptosis in cortex | Promotion of mitophagy mitigates CIRI | [76] |
| 2. Angiogenesis | |||||
| Primary neurons in the cortex and adult male SD rats | OGD/R model and tMCAO/R model | Upregulate NIX expression | 1. Increase ATP levels | Promotion of mitophagy mitigates CIRI | [77] |
| 2. Reduced neuronal cytotoxicity | |||||
| 3. Enhance mitochondrial content | |||||
| Wild-type male C57BL/6 adult mice | MCAO/R model | Inhibit the activation of the NLRP3 by promoting FUNDC1 expression | Reduced oxidative stress | Promotion of mitophagy mitigates CIRI | [78] |
Abbreviations: ATP, adenosine triphosphate; SD, sprague-dawley; BNIP3, BCL2/adenovirus E1B 19kDa
interacting protein 3; CaMKII, calcium/calmodulin-stimulated protein kinase II;
CIRI, cerebral ischemia-reperfusion injury; CK2
Ferroptosis, a type of programmed cell death, is marked by iron-dependent oxidative damage. Normally, cellular iron levels are dynamically balanced; however, excess iron can increase intracellular concentrations, leading to iron ion accumulation. This buildup induces membrane-lipid peroxidation and severe oxidative stress, culminating in cell death. Ferroptosis is morphologically distinguished by reduced mitochondrial size, increased membrane density, diminished or absent cristae, and eventual outer mitochondrial membrane rupture [79]. The regulation of ferroptosis involves complex biological pathways, including the activation of mitochondrial voltage-dependent anion channels and mitogen-activated protein kinases, an increase in endoplasmic reticulum stress, and the inhibition of cystine/glutamate antiporters. This process is linked to disruptions in iron metabolism, amino acid metabolism, lipid peroxidation, and oxidative damage [80]. Lipid peroxides (LPO) are key mediators in ferroptosis, with ROS and lipid peroxides being hallmark features [81], often associated with the downregulation of glutathione (GSH) and glutathione peroxidase 4 (GPX4) anti-oxidant systems. Studies have indicated that GPX4 [82], heat shock protein beta-1 (HspB1) [83], and nuclear factor erythroid 2-related factor 2 (Nrf2) [79] can mitigate ferroptosis by reducing iron uptake and inhibiting ROS production. Conversely, NADPH oxidase and p53 promote ferroptosis by enhancing ROS generation [84, 85, 86] (Fig. 2).
 Fig. 2.
Fig. 2.
Mechanism diagram of ferroptosis. The regulation of ferroptosis involves the activation of mitochondrial voltage-dependent anion channels and mitogen-activated protein kinases, an increase endoplasmic reticulum stress, and the inhibition of cystine/glutamate antiporters. FPN, ferroportin; GPX4, glutathione peroxidase 4; GR, glutathione reductase; GSH, glutathione; GSSG, Oxidized Glutathione; Keap1, kelch like ECH associated protein 1; Nrf2, nuclear factor erythroid 2-related factor 2; p62, sequestosome-1; ROS, reactive oxygen species; system Xc-, the xCT cystine glutamate antiporter; TfR1, transferrin receptor 1. Created the figure with Microsoft Office PowerPoint.
Iron overload is pivotal in triggering and advancing ferroptosis [87], directly elevating intracellular iron levels, and fostering lipid peroxidation (the core mechanism of ferroptosis). It influences ferroptosis through multiple pathways: (1) it has an impact on cell membrane stability [88] — iron overload exacerbates membrane-lipid peroxidation, leading to membrane rupture and cell death; (2) it promotes inflammatory response [89] — iron overload intensifies inflammatory stress, further contributing to cellular damage and death; (3) It has an effect on extracellular matrix stability [90] — iron overload disrupts extracellular matrix stability, affecting critical physiological processes such as cell invasion, migration, and angiogenesis, essential for ferroptosis progression. One study [91] has found that ferroptosis plays an important role in the occurrence and development of neurological conditions such as IS and CIRI.
Recent research has identified ferroptosis as a novel mechanism underlying CIRI, closely associated with brain cell death. Consequently, inhibiting ferroptosis has become a critical strategy for brain cell protection. Recent studies have highlighted the significant role of ferroptosis in the pathological processes of CIRI. During CIRI, ferroptosis is marked by iron overload, reduced GPX4 and GSH levels, and increased malondialdehyde (MDA) concentrations (a lipid peroxidation product) [24, 92]. CIRI induces excess iron ion influx into the brain parenchyma through a compromised BBB, promoting ROS production and lipid peroxidation via the Fenton reaction. Lipid peroxidation alters lipid chemical properties, impairing their ability to form functional cell membranes, resulting in membrane integrity loss and cell death, thus triggering ferroptosis-related cell death. MDA rapidly accumulates during this process, playing a vital role in cell death [93, 94]. Ferroptosis predominantly affects nerve cells during CIRI, exacerbating neuronal injury through mechanisms such as iron overload, excitotoxicity, antioxidant system disruption, and LPO. Numerous studies have shown that the three regulatory axes — Xc-/p53-GSH-GPXs [95, 96], ferroptosis suppressor protein 1-coenzyme Q10 (FSP1-CoQ10) [97, 98], and GTP cyclohydrolase 1-tetrahydrobiopterin-dihydrofolate reductase (GCH1-BH4-DHFR) [99] — are central to ferroptosis mechanisms and are integral components of the antioxidant system. Inhibiting ferroptosis via these key pathways may alleviate CIRI in patients. Additionally, ferroptosis activation contributes to neuronal dysfunction and death, intensifying post-ischemic brain injury.
Ferroptosis may worsen the pathological processes of CIRI by compromising BBB
integrity and enhancing inflammatory responses. The BBB is essential for
maintaining the stable environment of the central nervous system [100]. CIRI can
damage BBB structure and function, exacerbating inflammation in brain tissue and
contributing to neuronal cell death [101]. Therefore, protecting the BBB is
essential for preventing and treating CIRI. A recent study [102] has shown that
ginsenoside Rd inhibits CIRI-induced ferroptosis and prevents BBB leakage by
upregulating neuregulin 1 (NRG1), activating its tyrosine kinase receptor
Epidermal Growth Factor Receptor 4 (ErbB4), and stimulating the downstream
phosphatidylinositol 3-kinase-protein kinase B-mammalian target of rapamycin
(PI3K/Akt/mTOR) signaling pathway. This mechanism helps maintain BBB permeability
and mitigates the impact of CIRI on neurons. The inflammatory response is a
significant factor in BBB damage, and is often linked to iron overload.
Microglia, the primary immunomodulators of post-stroke inflammation, play a
pivotal role in this process [103]. Upon injury, infection, or inflammation,
microglia are modified into a macrophage-like state, releasing inflammatory
mediators and cytokines. The M1 and M2 phenotypes of microglia represent distinct
polarization states, with M1 microglia releasing a substantial amount of
inflammatory factors and neurotoxins after IS, leading to uncontrolled
inflammation and tissue damage [104]. M1-polarized microglia produce
pro-inflammatory cytokines such as interleukin (IL)-1
Nrf2 acts as a key regulator of cellular and systemic defenses against both endogenous and exogenous stressors. Its redox sensitivity allows it to modulate cellular redox balance and inflammatory responses [110]. A recent study [79] has shown that Nrf2 can enhance iron storage, decrease cellular iron absorption, and limit ROS production, thereby playing a key role in inhibiting ferroptosis. Activation of Nrf2 can mitigate ferroptosis, thus reducing CIRI damage and providing a neuroprotective effect.
During the initiation and progression of IS, Nrf2 is pivotal in several
signaling pathways, maintaining mitochondrial homeostasis, safeguarding the BBB,
and preventing ferroptosis. It achieves this by reducing oxidative stress,
inhibiting inflammatory responses, and regulating iron metabolism-related protein
expression. Nrf2 induces the expression of various antioxidant enzymes, such as
GPX4, glutathione S-transferase (GST), and superoxide dismutase (SOD), which,
collectively, reduce ROS production, thereby inhibiting ferroptosis. Through the
Nrf2/heme oxygenase-1 (HO-1) signaling pathway, Nrf2 enhances GPX4 levels, decreases the
accumulation of ROS, MDA, and Fe2+, boosts GSH levels, and increases SOD
activity, mitigating neuronal ferroptosis [111]. Moreover, Nrf2 diminishes
inflammatory responses and lowers the release of inflammatory factors. Research
has shown that it inhibits NLRP3 inflammasome activation and reduces
IL-1
In summary, Nrf2 is involved in regulating oxidative stress and modulating ferroptosis in CIRI. Its expression significantly affects the recovery and prognosis of IS injuries. Activation of Nrf2 enhances cellular antioxidant capacity, regulates iron metabolism, and inhibits inflammatory responses, providing new perspectives and potential therapeutic targets for CIRI treatment. Future research should delve deeper into the activation mechanisms of Nrf2 and the modulation of ferroptosis through Nrf2 activity, ultimately aiming to prevent and treat CIRI effectively. Studies of the mechanism regulating Nrf2-related signaling pathways that reduce ferroptosis in CIRI are summarized in Table 2 (Ref. [111, 120, 121, 122, 123, 124, 125, 126, 127, 128, 129, 130, 131, 132, 133, 134, 135, 136, 137, 138, 139, 140]).
| Cells or animals | Model | Target | Reference |
| Healthy male SD rats | MCAO/R model | Activating the Nrf2/HO-1 signaling pathway | [120] |
| Adult male SD rats | MCAO/R model | Activate Nrf2 and increase the protein expression of GPX4 and SLC7A11 | [121] |
| Male Kunming mice | MCAO/R model | Upregulate TAZ/Nrf2/HO-1 pathway | [122] |
| HT22 cells, V2 microglia cells and male SD rat | OGD/R model and MCAO/R model | Activate the Akt/GSK-3 |
[123] |
| PC12 cells and C57BL/6J mice | OGD/R model and MCAO/R model | Activate the Nrf2/GPX4 pathway | [124] |
| Adult male SD rats | MCAO/R model | Upregulate the expression of Nrf2, GPX4, and FPN | [125] |
| Primary culture cortical neuron cells and SD rats | OGD/R model and MCAO/R model | Regulate Keap1/Nrf2/HO-1 signaling pathway | [126] |
| HT22 cells and adult male SD rats | OGD/R model and MCAO/R model | Activate the Nrf2/GPX4 pathway | [127] |
| SH-SY5Y cells and male SPF-grade SD rats | OGD/R model and MCAO/R model | Enhance the anti-oxidant capacity of GPX4 through activation of Nrf2 signaling | [128] |
| PC12 cells and male C57BL/6 mice | OGD/R model and MCAO/R model | Upregulate the Nrf2, SLC7A11 and GPX4 protein expression | [129] |
| HT22 cells and male C57BC/6 mice | OGD/R model and MCAO/R model | Promote Nrf2 nuclear translocation and enhance intracellular antioxidant capacity of SLC7A11-GPX4 system | [130] |
| HT22 cells and adult male C57/BL6J mice | OGD/R model and MCAO/R model | Upregulate the expression of Nrf2, SLC7A11, GPX4, and LC3B protein | [131] |
| Adult male SD rats | MCAO/R model | Activate Nrf-2/HO-1/GPX4 signaling | [132] |
| SH-SY5Y cells and C57BL/6 mice | OGD/R model and MCAO/R model | Inhibit Akt/Nrf2 pathway | [133] |
| Male SD rats | MCAO/R model | Enhance Nrf2 nuclear translocation and regulation of ferroptosis-related proteins | [134] |
| Male SD rats | MCAO/R model | Increase Nrf2 nuclear translocation and the expression of SLC7A11 and GPX4 | [135] |
| Human umbilical vein endothelial cells and male SD rats | OGD/R model and tMCAO/R model | Regulate Nrf2/ARE/SLC7A11 signaling | [136] |
| HT22 cells and adult male C57/BL6J mice | OGD/R model and MCAO/R model | Upregulate the expression of Nrf2 and HO-1 and promoted the translocation of Nrf2 into the nucleus | [111] |
| HT22 cells and adult male SD rats | OGD/R model and MCAO/R model | Regulate the Nrf2/SLC7A11/GPX4 signaling pathway | [137] |
| SH-SY5Y cells and male C57BC/6 mice | OGD/R model and MCAO/R model | Activate Nrf2 pathway and promote nuclear translocation of Nrf2 | [138] |
| SH-SY5Y cells and male SD rats | OGD/R model and MCAO/R model | Activate the p62/Keap1/Nrf2 pathway | [139] |
| Male C57/BL6J mice | MCAO/R model | Regulate the Nrf2/GPX4 signaling pathway | [140] |
Abbreviations: Akt, protein kinase B; ARE, antioxidant response elements; FPN,
ferroportin; GPX4, glutathione peroxidase4; GSK-3
In CIRI, mitochondrial damage is a critical factor in the initiation of both mitophagy and ferroptosis [79]. Damaged mitochondria can be selectively removed via mitophagy or can lead to cell death through ferroptosis. As primary intracellular energy producers, mitochondria are major targets of oxidative stress, which facilitates the activation of mitophagy and contributes to ferroptosis onset. Oxidative stress is a significant trigger for mitophagy, releasing signaling molecules from compromised mitochondria, which activate autophagy-related pathways. Ferroptosis primarily relies on the accumulation of intracellular ROS and lipid peroxidation, with mitochondria being a major ROS source. Excessive ROS from abnormal mitochondrial metabolism disrupts redox homeostasis, triggering ferroptosis. Mitophagy reduces ROS production and accumulation by eliminating damaged mitochondria, thereby alleviating oxidative-stress-induced cellular damage to some extent. However, it may not completely prevent ROS accumulation and cell death during ferroptosis. This incomplete process can inhibit or destroy the antioxidant system, leading to ineffective ROS clearance and exacerbating ferroptosis [141]. In CIRI, ROS acts as a key link between mitophagy and ferroptosis, significantly influencing their occurrence, development, and interaction. The types of reactive oxygen species involved in both mitophagy and ferroptosis are summarized in Table 3 (Ref. [67, 68, 120, 135, 142, 143, 144, 145, 146, 147]).
| Mechanism | Type of reactive oxygen species | Reference |
|---|---|---|
| Mitophagy | superoxide anion | [142] |
| nitric oxide | [143] | |
| peroxynitrite | [67, 68, 144] | |
| Ferroptosis | superoxide anion | [145] |
| ozone | [135] | |
| nitric oxide | [120] | |
| peroxynitrite | [146] | |
| lipid peroxide | [147] |
Additionally, mitochondrial iron metabolism plays a role in regulating ferroptosis. Mitochondria, as primary organelles for iron metabolism, are central to this process. Increased intracellular iron content is associated with ferroptosis onset, and abnormalities in mitochondrial iron metabolism significantly affect its development. Excessive free iron in mitochondria can trigger ferroptosis by promoting lipid peroxidation through ferroautophagy, which releases free iron. Ferroautophagy also regulates the activity of mitochondrial Fe2+ transport proteins and ferritin levels, altering the abundance of free iron in mitochondria and influencing ferroptosis.
Dexmedetomidine (Dex), a widely used clinical
Given their shared targets and mechanisms, mitophagy and ferroptosis can influence and interact with each other through specific “bridges”. Mitophagy mitigates ferroptosis by clearing damaged mitochondria. During CIRI, ROS levels rise significantly. ROS not only directly damage mitochondria, triggering mitophagy, but also promote lipid metabolism and enhance lipid peroxidation, exacerbating ferroptosis. However, during CIRI, mitophagy activity may be inhibited, leading to the release of large amounts of iron ions from damaged mitochondria into the cytoplasm, which can trigger iron-dependent lipid peroxidation and promote ferroptosis [152, 153]. Excessively damaged mitochondria further activate mitophagy, reducing oxidative stress, decreasing iron ion accumulation, and removing ferroptosis-inducing factors, ultimately inhibiting ferroptosis. Additionally, one study has shown that NIP3 or PINK1-Parkin-mediated mitophagy can counteract cisplatin-induced damage to renal tubular epithelial cells through the ROS/HO-1/GPX4 signaling axis, thereby reducing ferroptosis [154].
Conversely, ferroptosis influences the activation and progression of mitophagy by altering mitochondrial function during CIRI. Significant changes in mitochondrial morphology occur during ferroptosis, including reduced mitochondrial size, increased membrane density, and loss of cristae; these changes have a direct impact on mitochondrial function. The excessive production of ROS during ferroptosis exacerbates mitochondrial damage, disrupting ATP production and redox balance. The diminished activity of GPX4, a key enzyme in the antioxidant system, leads to decreased cellular defense against lipid peroxidation and ROS accumulation [155], further compromising mitochondrial function. These pathways culminate in mitochondrial damage and dysfunction, subsequently activating the PINK1-Parkin pathway to induce mitophagy. In summary, a reciprocal regulatory mechanism exists between mitophagy and ferroptosis in response to CIRI. Mitophagy and ferroptosis may mutually regulate each other through shared signaling pathways. For instance, Nrf2 serves as both a key regulator of the antioxidant stress response and a modulator of iron metabolism. Future studies should focus on the role and mechanisms of Nrf2 in regulating these two forms of cell death.
CIRI is a complex pathological process that results in neuronal death and brain damage after the restoration of blood supply after ischemia. This process involves multiple processes, including oxidative stress, inflammatory responses, and apoptosis. Mitophagy and ferroptosis are two mechanisms closely associated with CIRI, both of which play significant roles in its progression; there exists a relationship between these mechanisms. The regulation of mitophagy activity can influence iron metabolism, and conversely, dysregulated iron levels can affect mitophagy. Therefore, further exploration of the interactions and regulatory mechanisms of mitophagy and ferroptosis in CIRI is crucial for understanding neuronal survival and functional recovery after such injury.
In CIRI, both energy depletion during ischemia, and oxidative stress after reperfusion, can lead to significant mitochondrial damage, which in turn may trigger apoptosis or necrosis. Mitophagy serves a protective role in brain tissue by removing damaged mitochondria, thereby reducing oxidative stress and apoptosis. However, excessive mitophagy may lead to the overzealous clearance of mitochondria, which would be detrimental to cell survival. Thus, the specific regulatory mechanisms of mitophagy and its dual effects in CIRI require further investigation. In CIRI, the metabolic disorder induced by ischemia, along with the oxidative stress response resulting from reperfusion, can significantly elevate the levels of free iron ions. This increase in iron ions subsequently induces ferroptosis because these ions promote lipid peroxidation. This leads to cell membrane damage and neuronal death. In CIRI, oxidative stress and the accumulation of iron ions are critical factors that trigger ferroptosis. Effective alleviation of CIRI can be achieved by reducing iron ion levels through the inhibition of ferroptosis-related pathways or by blocking lipid peroxidation with antioxidants. However, ferroptosis is a newly identified form of programmed cell death; currently, our understanding of the specific molecular mechanisms underlying ferroptosis in CIRI remains limited.
The conclusions of the present review have some limitations. Previous research [156] has demonstrated that the molecular mechanisms and pathophysiology of different subtypes of acute stroke vary. Mitophagy and ferroptosis can be inferred to differ among the various subtypes of IS and their corresponding CIRI species. However, there is currently a lack of differentiation among these subtypes, necessitating further in-depth studies. Additionally, although current research on the mechanisms of mitophagy and ferroptosis in CIRI has made some progress, it predominantly focuses on animal models and in vitro cell models. The validity of these results in humans and their potential for clinical application remain unclear, warranting further investigation. To address these issues, future studies could explore the spatiotemporal dynamic changes in the expression of mitophagy-related and ferroptosis-related genes and proteins at different stages of CIRI. Balancing the protective effects and potential adverse effects of mitophagy should be a primary focus of future research. It is crucial to clarify how related genes and proteins coordinate to regulate the occurrence of mitophagy and ferroptosis during CIRI and to fully understand their mechanisms of action. This understanding could pave the way for new strategies to prevent and treat CIRI and facilitate the development of related therapeutic agents.
This review elucidates the critical roles of mitophagy and ferroptosis in the pathogenesis of CIRI. Mitophagy serves as a protective mechanism by selectively removing damaged mitochondria, thereby reducing oxidative stress and inflammatory responses. Conversely, ferroptosis—driven by iron-dependent lipid peroxidation—exacerbates neuronal damage through ROS accumulation and BBB disruption. Their interplay centers on mitochondrial dysfunction and oxidative stress. The Nrf2 pathway emerges as a key regulator, balancing antioxidant responses and iron homeostasis. Despite therapeutic potential, challenges such as the dual regulatory effects of mitophagy and barriers to clinical translation persist. Future studies should explore the spatiotemporal dynamics of these processes across ischemic stroke subtypes and validate therapeutic interventions in human-relevant models to advance CIRI treatment paradigms.
YGM, XCW and XZ designed this review. YGM, XBW, YHL and JZ equally performed the literature search and wrote the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research was funded by “the National Famous Old Chinese Medicine Experts Inheritance Studio Construction Project, grant number [2022] No. 75” and “the Qilu BianCang Traditional Chinese Medicine Talent Cultivation Project, grant number [2024] No. 78”.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.