
- Academic Editors
†These authors contributed equally.
Lysine-Specific Demethylase 1A (Kdm1a) is the first discovered histone lysine-specific demethylase, and mutations in kdm1a have been detected in neurodevelopmental disorders. However, the effect of kdm1a on neurobehaviors and the underlying mechanisms remain largely unknown.
In this study, kdm1a deficient zebrafish were constructed using (clustered regularly interspaced short palindromic repeat) Clustered Regularly Interspaced Short Palindromic Repeats/CRISPRassociated protein 9 (CRISPR/Cas9) and the neurodevelopment was systematically assessed by a series of behavioral tests.
We found that kdm1a knockout zebrafish exhibited developmental toxicity and abnormal neurobehaviors, including locomotor abnormalities, and learning and memory deficits. Kdm1a deficiency suppressed central nervous system (CNS) neurogenesis in Tg (HuC:egfp) zebrafish, reduced motor neuron axon length in Tg (hb9:egfp) zebrafish and downregulated the expression of neurodevelopment related genes at 96 hours post fertilization (hpf). In addition, the expression of genes related to autophagy and apoptosis increased significantly in kdm1a knockout zebrafish.
These results indicated that kdm1a deficiency induced locomotor abnormalities and learning and memory deficits in zebrafish larvae accompanied by activation of autophagy and apoptosis. These findings indicate a key role of kdm1a in neurodevelopment, providing novel insights into the mechanisms underlying the neurodevelopmental disorders.
Lysine-Specific Demethylase 1A (Kdm1a) (also known as LSD1, or BHC110) is the first discovered histone lysine-specific demethylase, which is an amine oxidase histone demethylase. kdm1a maps to 1p36.12, which encodes a nuclear protein containing a Swi3p, Rsc8p, and Moira (SWIRM) domain, a flavin adenine dinucleotide (FAD)-binding motif, and an amine oxidase domain. Kdm1a is a component of several histone deacetylase complexes. It mono-methylates and di-methylates histone H3K4 or H3K9 via a FAD-dependent amine oxidation reaction [1, 2, 3]. Previous studies have shown that kdm1a plays an important role in a variety of physiological processes, such as the cell cycle, chromosome segregation, cell differentiation, cell proliferation, stem cell self-renewal, spermatogenesis, the epithelial-mesenchymal transition and tumorigenesis [4, 5, 6]. Kdm1a is also required for neurogenesis, and plays a role in neuron progenitor cell proliferation [7, 8, 9] and terminal differentiation [10, 11].
Mutations in kdm1a have been recently identified in a new neurodevelopmental disorder, which phenotypically resembles Kabuki syndrome but with distinctive facial features, skeletal anomalies and cognitive impairment [12, 13]. Additionally, several studies have shown that kdm1a is involved in neurological disorders. Christopher et al. [14] reported that deletion the kdm1a gene in adult mice leads to paralysis, along with widespread neuronal cell death in the hippocampus and cortex, and associated learning and memory deficits. However, studies on the potential molecular mechanisms of the neuronal damage mediated by loss of kdm1a are still limited.
A study has indicated that kdm1a is an essential regulator of autophagy [15]. Autophagy is accompanied by increases in microtubule-associated protein 1 light chain 3, lipidated LC3-II and cytosolic LC3-I (LC3II/LC3I) and decreases in sequestosome 1 (p62) [16]. Some studies have found kdm1a depletion triggers autophagy in neuroblastoma cells through the Sestrin 2-Mechanistic Target of Rapamycin Complex1(SESN2-MTORC1) pathway [17]. Autophagy is involved in neuronal damage [18]. Prostate cancer cells are suppressed by inducing apoptosis and autophagy can be induced with the specific kdm1a inhibitor N-[(1S,2R)-2-Phenylcyclopropyl]-1H-pyrrolo[2,3-b]pyridin-4-amine (NCL-1) [19]. Therefore, we hypothesized that loss of kdm1a may induce abnormal autophagy and apoptosis, which results in neurodevelopmental disorders.
Zebrafish (Danio rerio) are an ideal model for developmental and neurological studies due to their rapid external development, efficient reproduction, optical transparency and genetic similarities to humans [20]. Larval zebrafish begin to swim freely at 5 days post-fertilization (dpf) and the emergent patterns of development and movement are well-described [21]. Two transgenic (Tg) zebrafish models (HuC:egfp and hb9:egfp) provide visualization and analysis for neurogenesis and axonogenesis in vivo. Previous studies mostly focused on the effect of kdm1a on the developmental and behavioral characteristics in adult animal models [14, 22], while the effect during early life stages has been less studied. Hence, zebrafish were used to help elucidate the potential neurotoxicity induced by kdm1a deficiency and the potential mechanisms.
In this study, we established the inaugural kdm1a knockout zebrafish model, achieved through Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR-associated protein 9 (CRISPR/Cas9) genome editing. The kdm1a-deficient zebrafish larvae exhibited multiple behavioral abnormalities, such as locomotor abnormalities and learning and memory deficits during the early stages of development. Kdm1a deficiency also affected central nervous system (CNS) neurogenesis and reduced motor neuron axon length. Moreover, depleting kdm1a activated autophagy and apoptosis through abnormal gene transcription. These results strengthen our understanding of the role of kdm1a during early neurodevelopment, providing a potential new target for neurodevelopmental disorders in the future.
Rabbit anti-NeuN (1:1000, ab104225, Abcam, Cambridge, MA, USA), mouse anti-Microtubule-Associated Protein 2 (MAP2) (ab11267, Abcam, 1:1000), donkey polyclonal secondary antibody to rabbit IgG (1:1000, Alexa Fluor488, ab150061, Abcam), donkey polyclonal secondary antibody to mouse IgG (1:1000, Alexa Fluor594, ab150108, Abcam). Acridine orange (AO) stain (A9231) and tricaine methanesulfonate (MS-222) (E10521, 98% purity) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Hematoxylin and eosin (HE) (C0105M) and Nissl staining (C0117) were performed using commercial kits from the Beyotime Institute of Biotechnology (Shanghai, China). Trizol reagent (9109), reverse transcription reagent kits (RR037A), and SYBR-green RT-PCR kits (RR420A) were obtained from TaKaRa (TaKaRa, Dalian, Liaoning, China).
The Tg zebrafish (HuC:egfp andhb9:egfp) and wild-type
zebrafish (TU strain) were purchased from the model animal research center of
Nanjing University, China. All experiments were performed following the
Guidelines for Laboratory Animals. The zebrafish were maintained in a
recirculating culture system at 28.5 ℃ with a 10/14 h dark/light cycle according
to standard conditions. The water circulating in the system was filtered by
reverse osmosis (pH 7.5). The zebrafish were fed twice daily with brine shrimp.
The zebrafish larvae used in the behavioral tests were 5–10 days old. The fish
were randomly assigned to groups using a computer-generated randomization
sequence. The experimenter was unblinded to group allocation during data
collection and analysis to minimize bias. Blinding of the experimenter was not
feasible; however, steps were taken to mitigate bias, such as objective outcome
measures and independent assessors. After the experiment is completed, zebrafish larvae were euthanized at the designated time points by an overdose of tricaine methanesulfonate (MS-222, 300 mg/L) buffered with sodium bicarbonate (pH 7.0), followed by prolonged immersion (
The detailed procedure for zebrafish CRISPR/Cas9 editing was described previously [23]. The kdm1a target in this study was 5′-CAAAACCAAGCAGGACAACTT-3′. A solution containing 400 pg of Cas9 mRNA and 250 pg of gRNA was prepared for microinjection. Mutation sites were verified by comparing on the unaffected wild-type sequences (chimerism). To generate heterozygous kdm1a+/- mutants, chimeric founders were outcrossed to wild-type TU strain zebrafish for three consecutive generations. Then, kdm1a+/- males and kdm1a+/- females were crossed to obtain kdm1a-/- littermates.
Embryos were collected at 2 hours post-fertilization (hpf) and normally fertilized and developed embryos were selected for the subsequent experiment (n = 50 in each group). Hatching and survival rates were manually counted every 24 hpf. The malformation rate was quantified at 96 hpf, while heart rate was assessed at 48 hpf. After anesthetizing the fish in MS-222 (168 mg/L), the abnormally developing embryos during different periods were observed and captured by stereoscopic microscopy (SMZ18, Nikon, Tokyo, Japan). The fluorescence intensity of green fluorescent protein (GFP) in HuC:egfp zebrafish larvae (n = 10) and the axonal length of motor neurons in hb9:egfp zebrafish larvae (n = 10 in each group) were quantified using ImageJ software (version 1.53k; National Institutes of Health, Bethesda, MD, USA)
Zebrafish larvae were subjected to four behavioral tests, including a spontaneous locomotor activity test, the open field test, the mirror image attack test, and the Y-maze test. Previous studies reported that 12–15 larvae per group is suitable for assessing behavior [24]. To facilitate adaptation, zebrafish were allowed a 2-minute period for tank acclimation prior to experimentation. All tests were monitored and evaluated with the Zebralab ViewPoint system (version 3.90; manufactured by ViewPoint Life Sciences, Lyon, France) from 10 AM. to 5 PM. All experiments were performed at least three times independently.
A larval locomotion test was performed using a previously published method [25]. Zebrafish larvae (5 days post-fertilization [dpf], n = 12 in each group) were randomly selected from each group and added to a 24-well plate with a single animal in each well. Videos were recorded for 10 min by a camera on top of the tank. The swimming speed of the larvae was analyzed using ZebraLab software (version 5.10; manufactured by ViewPoint Life Sciences, Lyon, France), and the active times were quantified by the locomotor activity assay.
The open field test was conducted as described previously [26]. The experimental arena was partitioned automatically into 16 identical sectors, with the innermost four sectors designated as the central area. Zebrafish larvae (n = 12 in each group) were allowed to swim freely inside the tank for 15 min. The swimming distance and time spent in the central zone were calculated.
The mirror test was performed following the protocol of a previous study [27].
The transparent 5
The Y-maze task was conducted with 7–8 dpf fish to assess learning and memory
ability as described in previous studies [28]. The Y-maze was composed of three
arms at 120° to each other (6
To minimize potential experimenter bias during behavioral data collection and analysis, key experiments were conducted with the assistance of independent assessors who were not involved in the experimental treatment groups’ daily management and were blinded to the genotype/treatment conditions. For all larval behavioral tests, video recordings were scored automatically using automated tracking software where possible. For parameters requiring manual scoring, the videos were randomized and assessed by two independent researchers who were blinded to sample identity. Their scored results were then compared, and any discrepancies were re-evaluated jointly to reach a consensus. This approach ensured that the quantification of behavioral phenotypes was objective and unbiased.
To visualize apoptotic cells within 96 hpf larvae, acridine orange staining (AO) staining was carried out. Briefly, live larvae (n = 10 in each group) were cultured with AO solution (5 µg/mL) for 30 min in the dark at room temperature, and then washed three times. Stained larvae were photographed by a stereoscopic microscope following the manufacturer’s instructions (model Stemi 508; Carl Zeiss, Tokyo, Japan) after anesthesia (0.01% MS-222).
Zebrafish larvae (n = 10 in each group) were fixed in 4% paraformaldehyde solution (P0099, Beyotime) for 24 h and then transferred to a graded ethanol series. After dehydration in ethanol, the tissues were embedded in paraffinwax. Then the brain tissue blocks were sectioned at 5 µm thickness, and stained with HE for microscopic examination.
Paraffin sections were obtained by the method described above. The brain sections (n = 10 in each group) were dewaxed in a microwave with an antigen repair solution for 30 min. The slices were fixed in 4% paraformaldehyde at room temperature for 20 min and rinsed in water for 2 min. The treated samples were stained with Nissl staining solution for 5 min. The Nissl-positive cells were visualized under a light microscope (Product No.: C1791, MilliporeSigma, Burlington, MA, USA).
Paraffin sections were obtained by the method described above. The brain sections (n = 10 in each group) were dewaxed for 30 min in a microwave with an antigen repair solution, permeabilized for 5 min with 0.3% Triton X-100 (P0096, Beyotime) in PBS, and blocked for 30 min with 3% bovine serum albumin (ST023, Beyotime). The brain tissues were incubated with anti-MAP2 and anti-NeuN antibodies (1:200) overnight at 4 °C and stained with 4′,6-Diamidino-2-Phenylindole (DAPI) solution (C1006, Beyotime). Images were captured with a Nikon Eclipse Ti2 inverted fluorescence microscope (Serial N12345, Nikon Instruments Inc., Melville, NY, USA).
Total RNA was isolated from larvae sample (about 20 tails/group) with Trizol
reagent. First-strand cDNA was synthesized with Avian Myeloblastosis Virus (AMV) reverse transcriptase,
followed by SYBR Green-based qPCR analysis. Primer sequences for genes related to
neurodevelopment, autophagy and apoptosis are provided in Supplementary
Table 1. Gene expression levels were normalized to
Data are presented as mean
A 21-nucleotide guide RNA (gRNA) targeting exons 5–6 of the zebrafish kdm1a gene was designed to enable sequence-specific editing. To obtain kdm1a knockout (KO) zebrafish, the Cas9 mRNA and gRNA were injected into embryos. DNA sequencing of target-specific PCR products confirmed that the kdm1a targeted allele carried a deletion of one base, resulting in a frame shift mutation and premature translational termination (Fig. 1A and Supplementary Fig. 1). Homozygous kdm1a (kdm1a-/-) mutants were obtained from a heterozygous cross between kdm1a+/- males and kdm1a+/- females. Subsequently, homozygous kdm1a (kdm1a-/-) mutants were identified by DNA sequencing (Fig. 1B). Kdm1a-/- zebrafish exhibited a substantial decrease in kdm1a mRNA expression by 4 dpf (Fig. 1C). These results indicated that kdm1a KO zebrafish were successfully generated.
 Fig. 1.
Fig. 1.
Generation of kdm1a deficient zebrafish. (A) The
genomic and protein architecture of zebrafish kdm1a. The nucleotides in
black are gene sequences. The translated amino acid sequences are marked with red
words, and the amino acid which change resulting from frame shift mutations are
highlighted by yellow. (B) Sequence alignment of the WT and
kdm1a-/- zebrafish line, including the –1 bp deletion in
homozygotes. (C) Relative mRNA level of kdm1a in the kdm1a
deficient zebrafish larvae (n = 20 in each group). Data were presented as mean
To investigate the role of kdm1a in developmental abnormalities, we firstly analyzed the hatching rate, survival rate, heart rate and malformation rate. The hatching and survival rates of kdm1a-/- zebrafish decreased significantly after 48 hpf (Fig. 2A,B). While the malformation rate increased in kdm1a-/- zebrafish (Fig. 2C). No significant changes in the heart rate were observed in the kdm1a-/- group at 72 hpf (Fig. 2D). The typical morphological alterations mainly included pericardial edema (PE), yolk sac edema (YSE), curved body (CB) and low pigment (LP) (Fig. 2E).
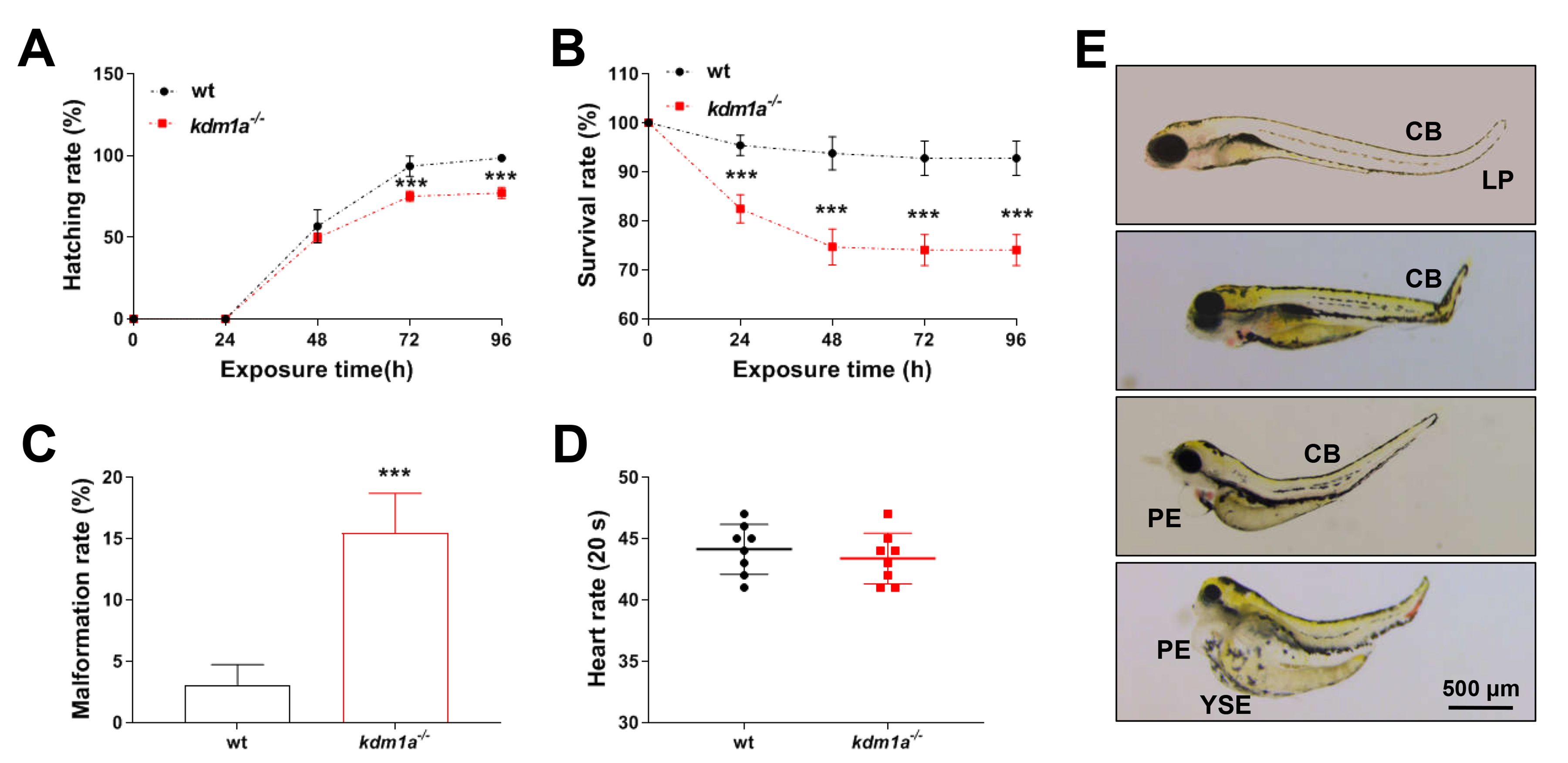 Fig. 2.
Fig. 2.
Developmental toxicity in kdm1a deficiency zebrafish
larvae. (A) Hatching rate. (B) Survival rate. (C) Malformation rate at 96 hpf.
(D) Heart rate. (E) Typically morphological alterations. The scale bar =
500 µm. PE, pericardial edema; YSE, yolk sac edema; CB, curved body; LP, low
pigment. Data were presented as mean
We used behavioral assays to analyze kdm1a deficiency in zebrafish and detected some abnormal behaviors. Spontaneous locomotor activity of individual larvae (5 dpf) was measured in a 24-well plate for 10 min. Active time decreased significantly in kdm1a-/- zebrafish (Supplementary Fig. 2A,B), indicating that the kdm1a deficiency impaired locomotion.
In the open field test, the typical locomotion tracking pattern illustrated differences in the exploration of the central and peripheral zones (Fig. 3A). Kdm1a-/- zebrafish exhibited less time and shorter distances in the periphery of the field (Fig. 3B,C), suggesting that kdm1a deficient zebrafish had a weakened ability to adapt to new environments.
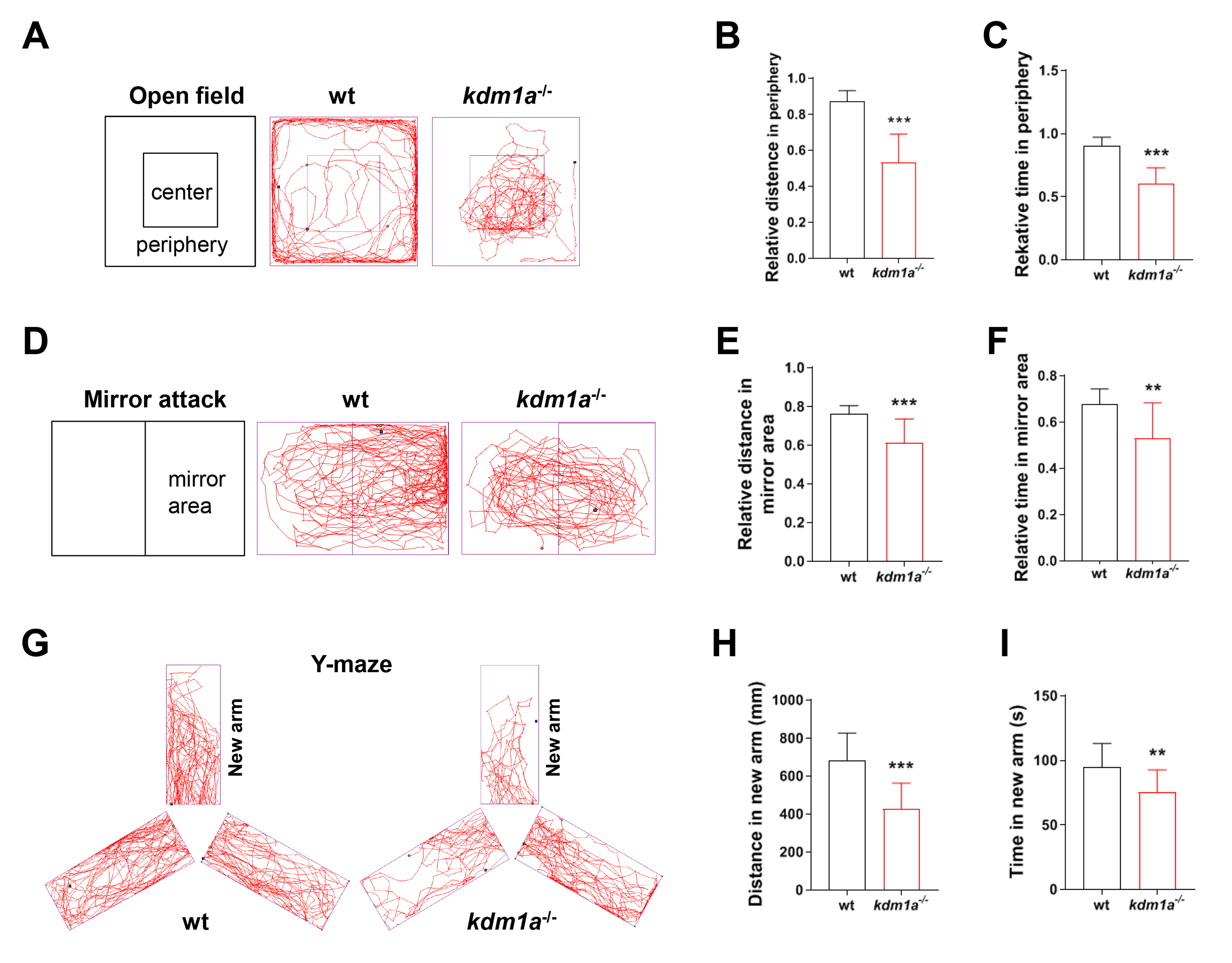 Fig. 3.
Fig. 3.
Neurobehavioral alteration in kdm1a deficiency
zebrafish larvae. (A) The motion trail recording, (B) total distance, (C) time
in the peripheral zone in the open field test at 120 hpf (15 min). (D) The motion
trail recording, (E) relative distance and (F) time in mirror area in the mirror
attack test at 120 hpf (15 min). (G) The motion trail recording, (H) distance and
(I) time in new arm in Y-maze test at 8 dpf (5 min). Data were presented as mean
The mirror attack test was used to study social behavior and response to novelty in zebrafish. The locomotion tracking patterns illustrated the differences between the mirror zone and the non-mirror zone during swimming traces (Fig. 3D). The time and distance traveled in the mirror area decreased significantly in kdm1a-/- zebrafish (Fig. 3E,F). In other words, the kdm1a-deficient zebrafish displayed less perception and interactive behavior, suggesting reduced cognitive ability.
To better understand the cognitive abilities of kdm1a-/- zebrafish, the Y-maze test was employed to analyze the time and distance in the novel arm. The locomotion tracking pattern illustrated the differences in swimming traces in Y-maze arms (Fig. 3G). As a result, significant decreases in the time and distance spent in the novel arm were observed in kdm1a-/- zebrafish (Fig. 3H,I), suggesting that cognitive ability, particularly learning and memory, was impaired in kdm1a-/- zebrafish.
To determine whether neurobehavioral dysfunction of kdm1a-/- zebrafish is closely associated with neurogenic impairment, we analyzed the brain structure and function of zebrafish. The HE and Nissl stained brain tissue revealed that the density of brain cells and Nissl bodies decreased in kdm1a-/- zebrafish (Fig. 4A,B). To investigate the effects of kdm1a on nervous system development, HuC:egfp and hb9:egfp zebrafish lines were used to determine the neurotoxic effects of kdm1a deficiency. As shown in Fig. 4C, kdm1a deficiency significantly decreased GFP intensity in the brain at 96 hpf. Similarly, the motor neuron axon length was significantly reduced in Tg (hb9:egfp) zebrafish (Fig. 4D). Moreover, immunofluorescent staining indicated significantly lower levels of NeuN and MAP2 (neuron markers) in kdm1a-/- zebrafish (Fig. 4E). The genes involved in early neurogenesis (neurod1, neurog1 and elavl3) and neural maturation (tuba1, gfap, gap43, and syn2a) were downregulated in kdm1a-/- zebrafish (Fig. 4F). These results illustrate that loss of kdm1a could induce significant neuronal impairment, which may be associated with abnormal behavior.
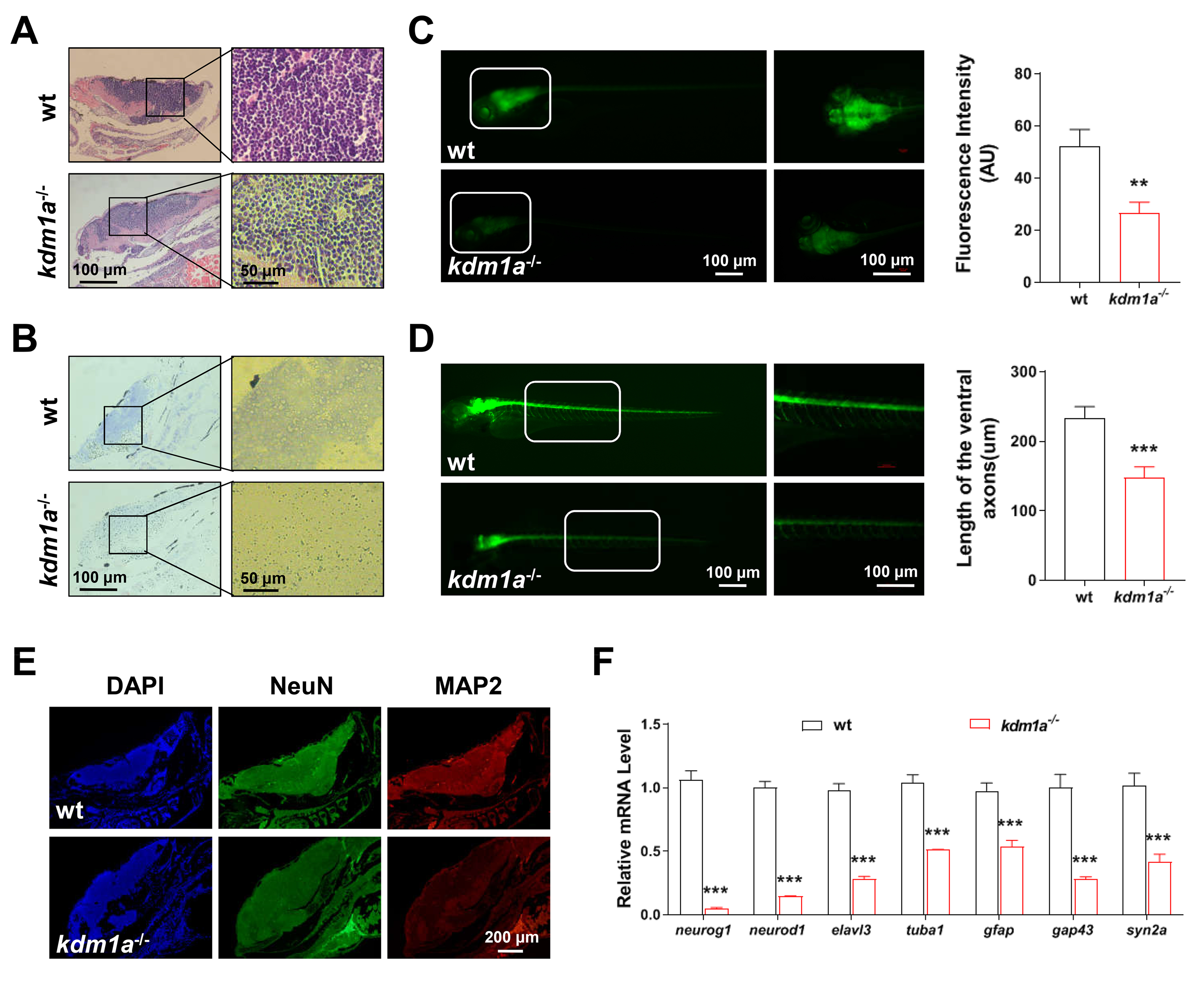 Fig. 4.
Fig. 4.
Kdm1a deficiency inhibited neurodevelopment in
zebrafish larvae. (A) HE and (B) Nissl staining of zebrafish brain in zebrafish
larvae (n = 10 in each group). Scale bar: 100 µm (left) and 50 µm
(right). (C) Representative fluorescence of neurogenesis in the CNS for whole
zebrafish (left) and a magnified view of the corresponding cerebral regions
(right) (n = 10 in each group). Scale bars: 100 µm. (D) Representative
fluorescence of motor neuron for whole zebrafish (left) and a magnified view of
the corresponding spinal regions (right) (n = 10 in each group). Scale bars: 100
µm. (E) Representative immune-stained images of MAP2 and NeuN in the brain
(n = 10 in each group). Scale bar: 200 µm. (F) Relative mRNA level of early
neurogenesis and neural maturation related genes (n = 20 in each group). Data
were presented as mean
To elucidate the mechanisms of neuronal damage in kdm1a-/- zebrafish, the expression of genes related to autophagy and apoptotic signaling was determined. As shown in Fig. 5A–G, the expression levels of caspase 3, caspase 8, caspase 9, lc3, and beclin1 increased, but the levels of p62 and bcl2/bax decreased in kdm1a-/- zebrafish. Kdm1a deficiency induced marked signs of neuronal apoptosis (Fig. 5H), indicating that loss of kdm1a triggered abnormal autophagy and apoptosis.
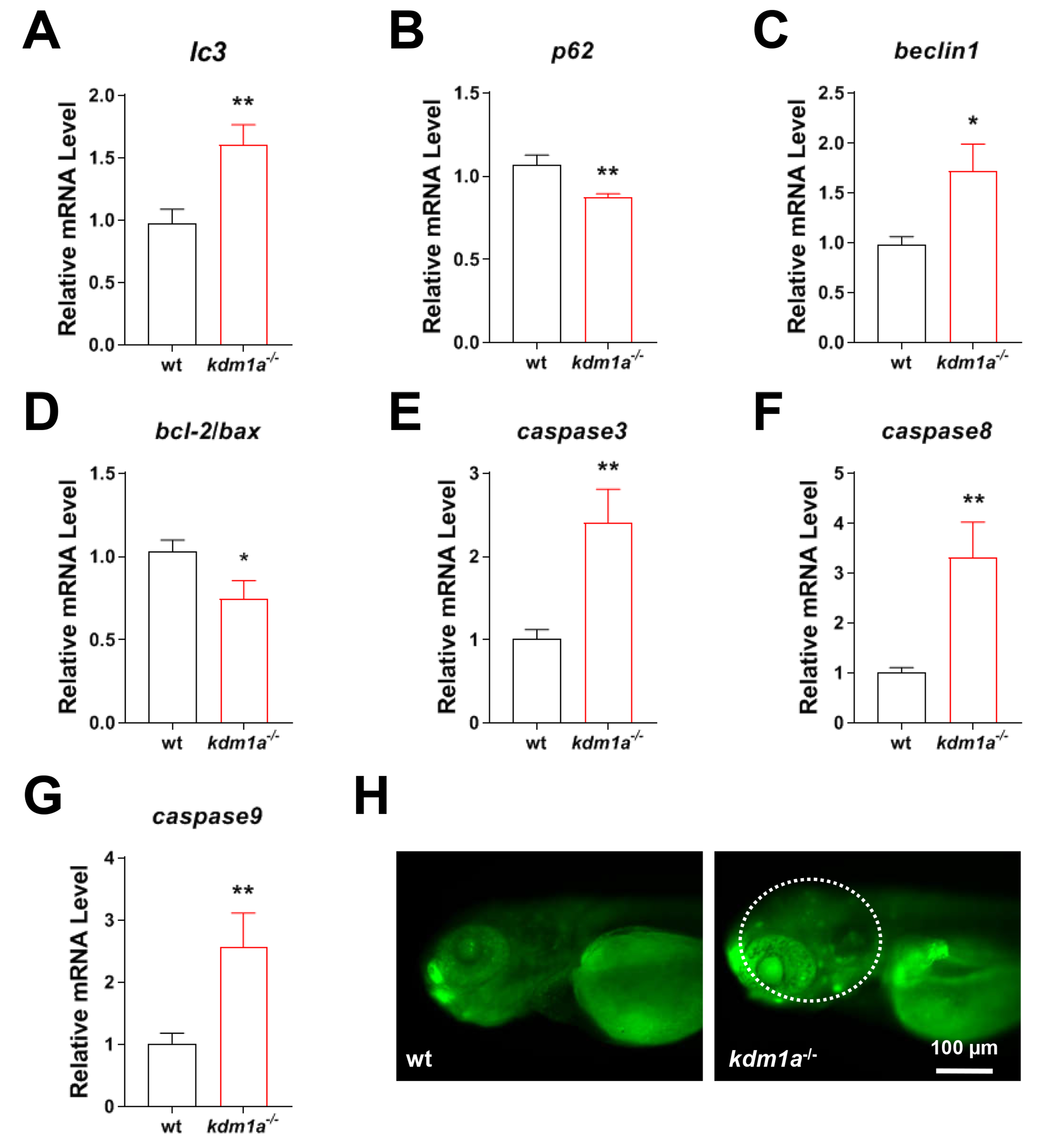 Fig. 5.
Fig. 5.
Kdm1a deficiency induced neuronal autophagy and
apoptosis in zebrafish larvae. Relative transcription activity of (A–C)
autophagy (lc3, p62 and beclin1) and (D–G) apoptosis
(bcl-2/bax, caspase 3, caspase 8 and
caspase 9). (H) Apoptotic cells in the brain. Scale bar: 100 µm.
Data were presented as mean
Neurodevelopmental disorders (NDDs), including syndromes characterized by abnormal CNS development, affect learning, cognition, emotion, and memory. Environmental and genetic factors contribute to neuronal impairment, resulting in NDDs [29]. Increasing evidence suggests that genetic factors play a major role in NDDs [30]. For example, a recent study reported that causal variants were identified in 36% of NDD individuals, and 23% of NDD individuals had uncertain significant variants [31]. KDM1A (also designated as LSD1), discovered in 2004, was the first histone demethylase to be identified. This enzyme utilizes FAD as a cofactor to catalyze the demethylation of histone marks, including H3K4me1/2 and H3K9me1/2 [32]. KDM1A is frequently overexpressed in a wide array of human cancers, such as acute myeloid leukemia (AML), prostate cancer, lung cancer, bladder cancer, lymphoid neoplasms, and breast cancer [33, 34, 35, 36]. Its oncogenic functions are mediated through diverse mechanisms: for instance, it regulates hematopoietic differentiation and promotes AML progression via H3K4 demethylation, while in breast cancer, it operates within the SIN3A/HDAC complex to support cell survival and tumorigenesis. Consequently, both genetic knockout and pharmacological inhibition of LSD1 have been demonstrated to effectively suppress tumor growth [37, 38, 39]. More recently, emerging evidence has revealed that KDM1A is not only implicated in tumorigenesis but also associated with NDDs. Pilotto et al. [40] found that three human patients with mutations in the kdm1a gene exhibited neurodevelopmental delay and mental retardation. Another study reported that deleting kdm1a in adult mutant mice induces severe paralysis, significantly reducing spatial learning and reference memory capacity, while hippocampal and cortical neurons appeared cell death [14]. Similarly, kdm1a knock-in (KI) mice exhibited short-term and long-term contextual fear memory as well as spatial memory deficits [22]. However, few studies have investigated the effects and mechanisms of kdm1a on neuronal development during the early life stages. Our study, which identifies a critical requirement for kdm1a in zebrafish neurogenesis, directly addresses this gap in knowledge and underscores the multifaceted nature of this epigenetic regulator.
In this study, we generated the first kdm1a-/- zebrafish using the CRISPR/Cas9 system and documented its morphological, behavioral and neurological characteristics. Potential off-target effects of the CRISPR/Cas9 system were mitigated by the use of a high-specificity sgRNA, designed to minimize sequence homology elsewhere in the genome, and by the genetic outcrossing of founders, which dilutes any random, off-target mutations. The consistent phenotypes observed across multiple independent mutant lines further support that they are the result of kdm1a loss-of-function specifically in the kdm1a-/- zebrafish. Previous studies focused on functional changes in kdm1a in adult mice, and kdm1a deficiency-induced developmental abnormalities have been less studied in embryos. In our study, kdm1a deficiency caused lower hatching and survival rates, and a higher malformation rate during early development. Kdm1a deficiency resulted in partial embryonic lethality and multiple morphological abnormalities during early development of zebrafish larvae. Interestingly, these observations, such as skeletal and cardiac malformations coincided with the clinical features reported in human kdm1a mutated patients [12, 13].
A series of behavioral tests have been applied in zebrafish, including assessments of locomotor activity, fear and anxious behavior, social interaction, novelty seeking, aggression and learning and memory [41]. In this study, we used some of these behavioral assays to analyze kdm1a deficiency in zebrafish and found some abnormal behaviors. The kdm1a-deficient zebrafish developed spontaneous locomotor deficit, suggesting that kdm1a deficiency decreases the ability to move, which coincides with the severe paralysis of kdm1a-deficient adult mice. In the open field test, kdm1a-/- zebrafish displayed significantly more exploratory behavior toward the center of the field, suggesting that kdm1a deficiency decreased the ability to perceive danger in a new environment, which somehow represents low intelligence [24]. The mirror attack test is typically used to study social behavior and the response to novelty in zebrafish. Interestingly, kdm1a-/- zebrafish interacted very little with familiar zebrafish (itself in the mirror). Zebrafish are interested in familiar fish, so they usually interact with the familiar opponent in the mirror. One of the possible reasons behind the abnormal behavior of kdm1a-/- zebrafish might be dysfunction in cognitive ability, particularly learning and memory deficits. More specifically, the zebrafish expressing exploratory activity in the open field test and interacting very little in the mirror test are thought to be related to cognitive deficits [42]. Therefore, we hypothesized that loss of kdm1a impairs the learning and memory ability in zebrafish and thus impacts cognition.
The Y-maze test was used to assess learning and memory, and the response to novelty in zebrafish was similar to that of rodents. The time and the distance traveled in the novel arm are the behavioral parameters in this test [43]. Zebrafish usually prefer the unexplored arm (novel arm). In this study, the kdm1a-/- zebrafish traveled a greater distance in the open arm and less in the novel arm, indicating memory deficits. These observations further suggest that loss of kdm1a induces learning and memory impairment. Taken together, kdm1a KO zebrafish exhibited motor deficits and intellectual disabilities, which were consistent with Christopher et al.’s findings [14] that kdm1a deficiency causes paralysis and learning and memory deficits in adult mice. Few studies have investigated the role of kdm1a during early neurodevelopment. These behavioral phenotypes provide deeper insight into kdm1a-KO, indicating the important role of kdm1a in neurodevelopmental behaviors in zebrafish larvae.
Neurobehavioral deficits are closely associated with neurogenic disruption. Interestingly, the HE and Nissl-stained sections revealed that kdm1a-/- zebrafish had significantly fewer neuronal cells than the control group, suggesting that loss of kdm1a causes neurogenic impairment, which may lead to morphological and behavioral abnormalities. The results of this study follow previous findings demonstrating that neural cell death assessed by Nissl staining has a detrimental effect on animal behavior [44].
The neurobehavioral changes in zebrafish larvae are closely related to their neurogenetic or axonogenetic disorders [20]. To further validate this assumption, the effects of kdm1a on CNS and motor neuron development were evaluated using HuC:egfp and hb9:egfp transgenic zebrafish. In HuC:egfp transgenic zebrafish, GFP was integrated into the promoter sequence of the elavl3 gene, which encodes the neuron-specific RNA-binding protein HuC. HuC is one of the earliest neuronal markers in zebrafish and is expressed in the CNS [45]. In the hb9:egfp zebrafish, GFP is specifically localized to motor neurons under the regulatory control of the hb9 gene, a key regulator essential for motor neuron development [46]. Consistent with the neurobehavioral changes, kdm1a deficiency significantly reduced GFP intensity in the brain of HuC:egfp transgenic zebrafish at 72 hpf and inhibited motor neuron axon growth in hb9:egfp zebrafish.
Neuronal nuclei (NeuN) and microtubule-associated protein 2 (MAP2) are two neuron-specific proteins. Due to conservation among species and their stable expression during specific developmental stages, NeuN and MAP2 are reliable, conserved markers of mature neurons [47]. MAP2 and NeuN expression levels have been thought to indicate neuronal death or loss [48]. In this study, the brightness of the fluorescent NeuN and MAP2 staining decreased, indicating neuronal damage. In addition, we measured the mRNA expression of neurodevelopmental genes (elavl3, neurog1, neurod1, tuba1, gap43, gfap and syn2a) to verify the neurotoxic effect of kdm1a. Elavl3, neurog1 and neurod1 serve as biomarkers for early neurogenesis in zebrafish [49], whereas gap43, gfap and syn2a are linked to neural maturation, axonal growth and neurotransmitter secretion, particularly synaptic functions [20]. In our study, loss of kdm1a downregulated these genes, which further demonstrated that loss of kdm1a exerted direct effects on neurogenetic and motor neuron axonogenetic injury, thereby changing the neurobehaviors of zebrafish larvae.
The mechanisms of kdm1a-induced damage on motor neuron axonogenesis and neurogenesis are largely unexplored. Recent studies have demonstrated that Autophagy has been recently reported to participate in the development process of NDDs [50, 51]. Autophagy is a conserved self-destructive process used to remove damaged organelles and proteins via lysosomal degradation. Autophagy plays a crucial role in the organogenesis in zebrafish, including neurogenesis. Multiple lines of evidence point to kdm1a as an essential regulator of autophagy [52]. Some studies have shown that kdm1a affects autophagy by epigenetically modifying the expression of some proteins [53]. Moreover, kdm1a may also directly affect proteins involved in autophagy, such as P62 [54]. In this study, we observed the changes in autophagy-related molecules (P62, Beclin1 and LC3) and found that a deficiency of kdm1a increased the expression of beclin1, lc3 and decreased the expression of p62. Our data demonstrated that a lack of kdm1a induced excessive autophagy in zebrafish larvae.
More importantly, excessive autophagy is a potential pathway to induce neuronal apoptosis [55]. For example, myclobutanil exposure causes excessive autophagy and neuronal apoptosis, leading to developmental neurotoxicity in zebrafish [56]. Therefore, autophagy is an inducer of apoptosis by activating the mitochondrial apoptosis pathway in zebrafish [57]. In the present study, we further investigated the changes in mitochondrial apoptosis-related molecules (bcl-2, bax, caspase3, caspase9 and caspase8) and found that kdm1a deficiency increased the expression of caspase3, caspase9 and caspase 8, and decreased the expression of bcl2/bax, resulting in the accumulation of apoptotic cells in the brain of zebrafish larvae. Collectively, our data demonstrated that a lack of kdm1a induced hyperactive autophagy and neuronal apoptosis, coinciding with aberrant behaviors and neurodevelopment in zebrafish larvae. This suggests that the dysregulation of these cellular processes may be a significant contributor to the observed neurotoxicity.
Nevertheless, this study has limitations as we cannot directly confirm the loss of KDM1A protein at the biochemical level due to the lack of a validated antibody against zebrafish KDM1A, future efforts will focus on obtaining or generating a specific antibody to definitively confirm protein ablation. Due to the absence of kdm1a overexpression in both wild-type and kdm1a-deficient zebrafish models, conducting gain-of-function and rescue experiments in future work will be essential to further clarify the precise functional contributions of kdm1a, its influence on neural development, and the molecular mechanisms involved. While our findings link kdm1a loss to autophagy/apoptosis activation and neuronal defects, the exact mechanism requires further investigation. Future studies should use autophagy and apoptosis inhibitors in zebrafish to determine whether suppressing these pathways rescues the neurodevelopmental phenotypes.
In summary, the present study demonstrates that kdm1a deficiency leads to excessive autophagy and neuronal apoptosis, which are likely responsible for the impairments in neurogenesis, motor axon outgrowth, and learning and memory in zebrafish larvae. These findings establish a critical link between kdm1a dysfunction and behavioral abnormalities relevant to NDDs, advancing our mechanistic understanding of neurodevelopmental and axonal pathogenesis mediated by kdm1a dysregulation. Our study provides new insight into developing potential therapeutic strategies for autophagy and apoptosis to limit the pathogenesis of NDDs.
The data that support the findings of this study are available from the corresponding author with reasonable request.
LZ, Conceptualization, Data curation, Visualization, Original draft, Writing-Reviewing and Editing. JW, Conceptualization, Data curation, Methodology, Software. MY, Methodology, data analysis. QX, Methodology, data analysis. QH, Methodology, data analysis. MY, Methodology, data analysis. JZ, Methodology, Data curation, Writing-Reviewing and Editing, Validation, Funding acquisition. XC, Conceptualization, Supervision, Writing-Reviewing and Editing, Funding acquisition. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
All experiments complies with American Veterinary Medical Association (AVMA) guidelines for euthanasia of zebrafish and adhered of the Animal Care Committee of Nanjing Medical University (IACUC-2205028).
Not applicable.
This work was supported by the National Natural Science Foundation of China [82103877], the Natural Science Foundation of Jiangsu Province [BK20211017].
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/JIN44394.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.