
 , Esra Aslan 2
, Esra Aslan 21 Department of Psychiatry, Health Science University, Adana City Health Research and Application Center, 01230 Adana, Turkey
2 Department of Psychiatry, Aksaray University, Aksaray Training and Research Hospital, 68200 Aksaray, Turkey
Abstract
The feeling of emotional tension, restlessness, pressure, and inability to relax is referred to as psychological stress. Although it is unclear how psychological stress affects neurobiological processes, several factors are thought to be involved, including central and peripheral neuroinflammation, structural degeneration in the prefrontal cortex and hippocampus, alterations in fear neurocircuitry, and neuroplasticity. Aside from data relating cognitive impairment to chronic low-grade inflammatory stress, there is growing evidence linking mental stress, oxidative stress, and systemic inflammation to the development of psychological disorders. After chronic and acute illnesses, insomnia, depression, anxiety, posttraumatic stress disorder, and cognitive impairment were reported. Cognitive impairment is exacerbated by systemic and central inflammatory processes. There is uncertainty about the potential mechanisms causing these symptoms, although they are likely complex, with systemic inflammation playing a significant role. Therefore, this review aims to investigate the role of inflammation in stress-induced cognitive impairment. Depicting the inflammatory mechanisms of cognitive impairment is critical for understanding and treating illnesses, such as chronic stress exposure and anxiety disorders.
Keywords
- neuroinflammation
- cognition
- cognitive impairment
- stress
- inflammation
Stress is a condition of impaired homeostasis that occurs in response to an actual or perceived threat. Stress responses help us adapt to our environment and respond appropriately to threats [1, 2]. However, the stress response mechanism that protects the organism must also be terminated. A prolonged alarm condition would cause the system to become unstable. Stress has a negative impact on various aspects of brain function when it is sustained or repeated. In particular, uncontrollable, prolonged stress leads to neuropsychiatric disorders [3, 4]. Moreover, chronic stress is associated with life-threatening conditions, such as cancer, insulin sensitivity, and cardiovascular disease [5, 6].
This review focuses on psychological stress. Emotional tension, mental discomfort, and pressure are symptoms of psychological stress. It may be caused by work stress, exposure to traumatic events, disappointment, unhappiness in marriage, confronting unanticipated negative situations, failure to meet basic needs, and caregiving burden. The psychological stress mentioned in this review is different from mechanical stress. A stress response is triggered when tissue integrity is disrupted, as in bone fractures and burns. This is referred to as mechanical stress. Mechanical stress causes processes, such as psychological stress, and is associated with oxidative stress. However, it was not considered in this review [7].
Traumatic stress affects neurocognitive functions. One of the best examples of how stress affects cognitive functions is posttraumatic stress disorder (PTSD). If PTSD is revealed, neurocognitive functions are impaired with deficiencies in attention, executive function, and memory [8]. Moreover, when we review the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5) diagnostic criteria, negative cognitive alterations and difficulties concentrating are some of the criteria for the diagnosis of PTSD [9]. Inflammatory diseases also affect cognitive functions. The relationship between inflammatory diseases and impaired cognition has been investigated for a long time. Some immune modulators, such as cytokines and microglia that increase inflammation, affect cognitive functions [10, 11]. In this review, the role of inflammation in stress-induced cognitive impairment is discussed in the following order: stress-induced subclinical inflammation, followed by inflammation-induced cognitive impairment.
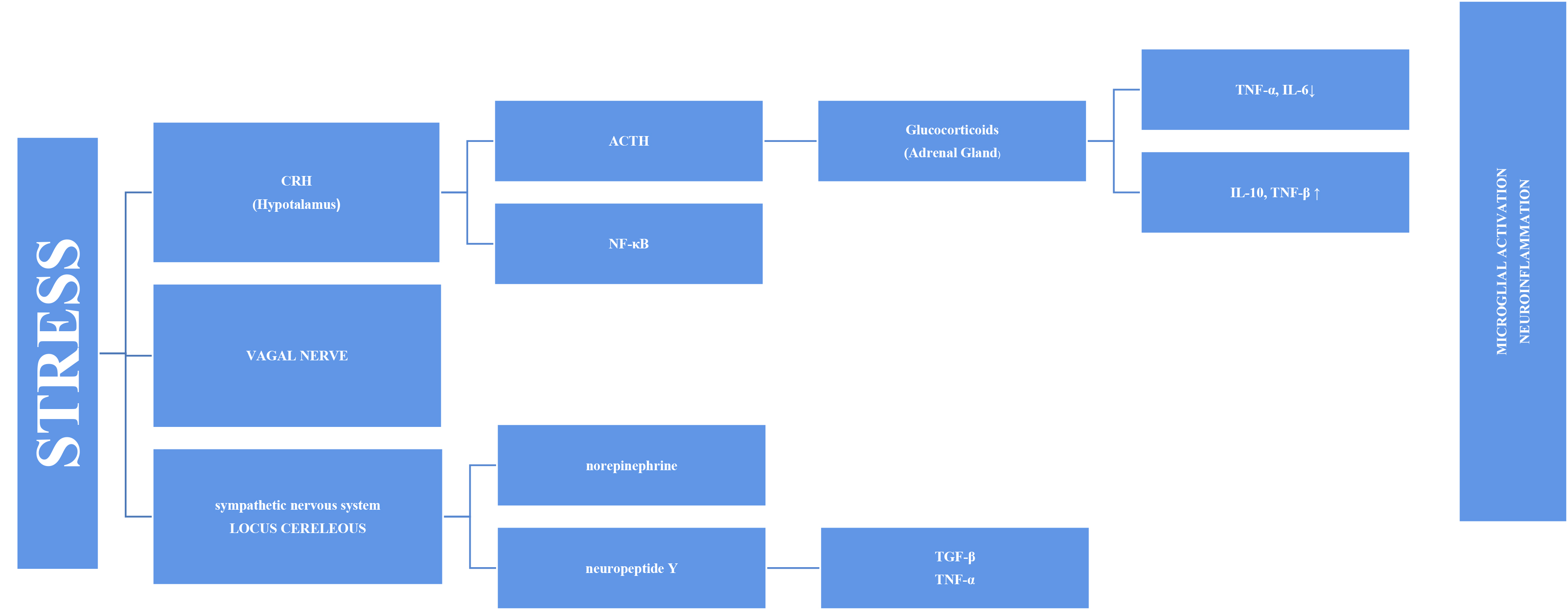
Stress has long been thought to be associated with inflammation because depressive symptoms are common in individuals with autoimmune and inflammatory diseases, such as diabetes and fibromyalgia. An increasing number of studies on this subject have been conducted [12]. Inflammation and increased pro-inflammatory cytokines have been associated with PTSD, depression, and various psychopathologies [13, 14]. This has prompted more research into the cause-and-effect relationship and mechanism. Stress systems transmit threats throughout the body and stimulate inflammatory mechanisms [8]. The stress response adapts quickly to risks, such as increased blood sugar, blood pressure, and heart rate. However, the systems also stimulate the inflammatory response by producing inflammatory cytokines in the blood via the sympathetic branch of the autonomic nervous system (Fig. 1). This initial response is mediated by the hypothalamic–pituitary–adrenal (HPA) axis and the final hormone cortisol on this axis [15]. This acute stress is called “subthreshold” because it does not trigger increased inflammatory responses [16]. However, this article discusses other factors that may cause long-term sensitivity. Cortisol has a wide range of effects on the body and brain and is assumed to play essential roles in daily cognitive and behavioral functions. Chronic stress, such as depression, causes glucocorticoids resistance in the HPA axis, enabling pro-inflammatory signaling pathways to bypass the typical feedback inhibition [17]. Cortisol has various effects on the brain. One of these is increased cognitive deterioration associated with improved neuroinflammatory processes. Cortisol over production primes microglia for an enhanced pro-inflammatory response to stress, which boosts levels of neurotoxic cytokines [18].
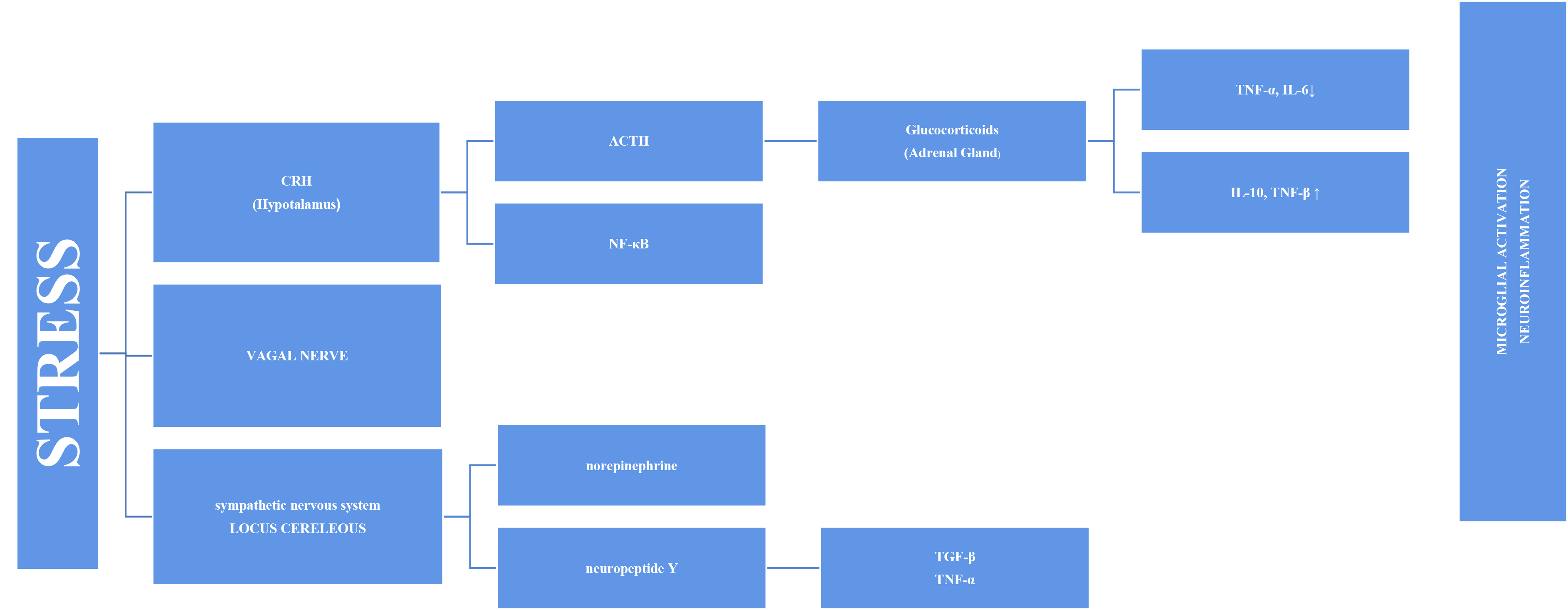 Fig. 1.
Fig. 1.Representation of microglial activation via the SNS and HPA axis
in a diagrammatic form. SNS, sympathetic nervous system; HPA, hypothalamic–pituitary–adrenal; TNF-
The vagus nerve is another mechanism in the link between inflammation and
stress. The circuits underlying the symptoms of stress-related psychiatric
diseases are regulated by the vagus nerve. Vagus nerve stimulation binds to the
alpa-7 subunit of the cholinergic receptor, activating T cells that produce
acetylcholine and inhibiting nuclear factor kappa B (NF-
The stress response involves the sympathetic system as well as the HPA axis, in the axis of “nervous, endocrine and immune” mechanisms. The adrenal medulla secretes more norepinephrine (NE) and epinephrine (E) into the bloodstream as a result of the quick reaction brought on by S-Adenosil metiyonin (SAM) activation. Sympathetic nerves also secrete more NE, which raises NE levels in the brain [24]. The organism becomes more vigilant and responds quickly to environmental cues when NE is released. Chronic stress causes locus coeruleus (LC) neurons to become hyperactive, which may be a factor in stress-related depression disorders. Chronic stress may have a mediating effect on microglia through catecholamines. According to recent research, microglia probably take part in the neuroinflammatory response that results from stress, partly via noradrenergic transmission [25]. It has been proposed that NE can upregulate the transcription of the proinflammatory immune response genes interleukin-1 (IL-1), tumor necrosis factor (TNF), and interleukin-6 (IL-6) and decrease the transcription of antiviral type I interferon (IFN) genes [26, 27].
As a result, chronic subclinical inflammation causes a sustained systemic increase in inflammatory immune mediators. That sustained inflammation is associated with cell damage. Various transcription factors or inflammasomes may play a role in this case, as explained in the following sections.
Combined with cortisol release, the adrenal glands respond by releasing glucocorticoids and catecholamines. Catecholamines (e.g., norepinephrine) are released when the sympathetic nervous system is activated and functions directly on the primary and secondary lymphoid tissues. Glucocorticoids and catecholamines speed up monocyte synthesis, maturation, and release into circulation [16]. These monocytes enter the brain and become enriched in the hippocampus, prefrontal cortex, and amygdala. However, after microglial activation, the M1 phenotype produces pro-inflammatory cytokines. Therefore, it is plausible that the inflammatory reaction caused by microglial activation may be a factor in brain pathology including cognitive decline [28]. Moroever, activated microglia is able to secrete large amounts of glutamate and increased glutamate release may contribute to excitotoxicity [29]. Extremely motile, microglial processes directly connect with synapses that rely on continuing neuronal activity in part. Recent data demonstrated the critical role NE signaling plays in mediating these dynamic relationships [25, 30]. Thus, chronic stress causes an increase in NE signaling. This implies that chronic stress-induced microglial overactivation may result in a reduction in microglia-neuron connections, which could have a negative impact on synaptic plasticity and possibly neural connectivity [25]. Another reason why cognitive functions in the brain are affected may be inflammasomes. Inflammasomes are a multiprotein complex that intensifies inflammatory responses against endogenous activators, not only in response to pathogens but also in response to psychological stress [31]. The most extensively studied inflammatory complex of these is the Nod-like receptor pyrin domain-containing 3 (NLRP3) inflammasome. Research has demonstrated that autophagy and inflammasomes are mutually controlled. The intracellular process of autophagy is critical for both the removal of intracellular pathogens and the recycling of damaged proteins and organelles. Through hyperinflammation and excessive activation of inflammatory cells, autophagy failure can result in inflammatory disorders [32, 33].
During M1 activation, microglial cells secrete pro-inflammatory molecules, such
as interleukin-1
2.1.1.1 Interleukin-1
IL-1
2.1.1.2 Interleukin-8 (IL-8)
IL-8 is a pro-inflammatory cytokine produced by various cell types, notably macrophages and microglia. Its pro-inflammatory activities include promoting neutrophil adhesion, chemotaxis, and lysosomal discharge [37]. Clinically, IL-8 is essential in various inflammatory and autoimmune disorders. There have been reports of increased IL-8 production in the peripheral systems of patients suffering from various inflammatory diseases, autoimmune disorders, infections, and cancers [38, 39]. Notably, IL-8 appears early in the inflammatory cascade and may persist for extended periods, unlike most other inflammatory cytokines, typically produced and cleared within a few hours [40]. This unique persistence suggests that IL-8 may have a role in chronic inflammatory changes, particularly in neurodegenerative and neuropsychological changes in the brain [41].
2.1.1.3 Interleukin-6 (IL-6)
IL-6 is a pro-inflammatory cytokine that stimulates the production of inflammatory acute-phase proteins [42]. These proteins majorly affect vascular permeability, lymphocyte activation, and antibody synthesis. IL-6 was the predominant cytokine in mouse models that can only be induced by acute stress [43]. This stress-induced cytokine is produced from brown adipocytes via the beta-3-adrenergic receptor. One of its functions is to mediate hyperglycemia in the “fight or flight” response via hepatic gluconeogenesis. The cost of this adaptation is increased mortality from a subsequent inflammatory challenge [44]. Within the central nervous system (CNS), IL-6 can function as an indirect immunosuppressive factor by strongly stimulating the pituitary–adrenal axis, producing adrenocorticotropic hormones [45]. As a result of this hormone release, the adrenal gland increases the synthesis of glucocorticoids.
2.1.1.4 Tumor necrosis factor-
TNF-
2.1.2.1 Interleukin-4 (IL-4)
In numerous experimental CNS disease models, interleukin-4 has been suggested as a neuroprotective and regeneration-inducing agent. While most studies have focused on myeloid cells, research on the neuronal IL-4 receptor (IL-4R) signaling pathway suggests that it directly affects neurons. The pleiotropic cytokine IL-4 is important in modulating immune responses. It has an effect when it interacts with high-affinity receptor complexes found in hematopoietic and non-hematopoietic cells [49].
2.1.2.2 Interleukin-10 (IL-10)
Interleukin-10, a well-known anti-inflammatory cytokine, is essential in modulating multiple immune functions in response to inflammation. Its significance becomes apparent during the resolution phase. There is an increase in IL-10 expression inside the brain in cases of CNS pathology. This up regulation helps preserve neuronal and glial cells while suppressing inflammatory responses via various signaling pathways [50]. IL-10, initially known as a cytokine synthesis inhibitory factor, inhibits the production of pro-inflammatory cytokines and reduces inflammation by reducing cytokine receptor expression and preventing their activation [50].
2.1.2.3 Transforming Growth Factor-
Distinguishing itself from other cytokines, the transforming growth
factor-
The consequences of this subclinical inflammation stimulated by long-term psychological stress will also be seen in the brain. Significant evidence points to the role of inflammatory cytokines in directing the brain’s inflammatory responses, eventually resulting in injury to its cellular components in neurodegenerative disorders, such as cognitive impairments [53].
The hippocampus is a critical brain region that is the focal point for learning and memory processes. Dysfunction of the hippocampus is important in altering cognitive abilities. Early-onset hippocampal inflammation is a hallmark event in the progression of neurodegenerative disorders, potentially leading to long-term changes in brain function during its developmental phases. Prolonged changes in hippocampal function caused by inflammation significantly impact brain excitability, which is associated with various neurological dysfunctions [54]. There is a precursor form of IL-18 in brain neural cells, and its absence causes hippocampal dysfunction and a depressive-like phenotype. As a result, IL-18 has been associated with immunological function, psychiatric traits, including depression, and neurological function [55].
The subsequent content introduces several significant theories regarding the involvement of key molecules in the dynamics of psychological stress.
A member of the neurotrophic factor family found in the brain is called brain-derived neurotrophic factor (BDNF). It is associated with psychological resilience. It remains one of the most studied molecules in psychiatry and is a common hereditary risk locus for mental disease. Stress, a risk factor for several psychopathologies, targets BDNF in disease-related brain circuits and regions. BDNF plays a complex and underappreciated role in each of these processes as a target and regulator of stress hormone transmission in the brain. Stress hormones affect brain homeostasis by interacting with endogenous BDNF–Tropomyosin receptor kinase B (TrkB) signaling [56]. Additionally, BDNF is acutely recruited by glucocorticoid signaling to promote the development of fear memory [57]. BDNF was found to be required for the pro-neurogenesis effects of IL-4-induced microglia. Microglia activated by IL-4 in the hippocampus have been shown to help protect against depressive-like symptoms by inducing BDNF-dependent neurogenesis in response to chronic stress [58, 59]. In animal studies, pubertal trauma has been associated with increased corticosterone and reduced BDNF levels in the dorsal hippocampus [55]. According to Ferrer et al. [60], single nucleotide polymorphisms (SNPs), methylation status, and interactions with modifying variables in the BDNF gene can affect cognition.
Brain-derived neurotrophic factor alters the synaptic plasticity. Synaptic
plasticity, or the ability of brain connections to become stronger or weaker over
time, underlies cognitive functions, such as memory development and recall [61].
BDNF controls memory processing on several molecular levels, includingcation
(Na
The precise synaptic connections formed between inhibitory interneurons and their excitatory counterparts are critical to adequately operating neural circuits. Inhibitory interneurons, which account for 10%–20% of the neuronal population, are responsible for modulating the activities of excitatory neurons [66]. Parvalbumin (PV) interneurons, a predominant category of inhibitory interneurons, are critical in maintaining the balance of excitation and inhibition within neural circuits [67]. Research showed that inflammation reduces the expression of PV interneurons, eventually leading to PV interneuron dysfunction and cognitive impairment [68].
Sirtuin 1 (SIRT1), a well-known member of the Sirtuin family, is essential for
various cellular physiological and biochemical operations, including managing
aging-related senescence and inflammation [69]. SIRT1 deficiency appears to have
a role in developing cognitive abnormalities in the context of neurodegenerative
diseases within microglia, possibly through epigenetic modulation of
IL-1
SIRT1 is important in managing the biological condition known as oxidative
stress. This vital function includes regulating key transcription factors, such
as nuclear erythroid factor 2-related factor 2 (Nrf2), which causes the
transcription of antioxidant enzymes, influencing the cellular redox equilibrium.
As a redox-responsive transcription factor, Nrf2 acts as a mediator for various
cytoprotective [70] elements, including heme oxygenase-1 (HO-1) and antioxidant
enzymes, such as superoxide dismutase. In addition, through deacetylation, SIRT1
inhibits the mammalian target of rapamycin (mTOR) and triggers autophagy. Thus,
autophagy activities break down and remove
One of the most prevalent peptides in the body is Neuropeptid Y (NPY). Its
effects on the peripheral nervous system and central nervous system are
extensive. The brain’s hypothalamus, septum, nucleus accumbens, periaqueductal
gray, and locus coeruleus have particularly high levels of NPY expression.
Numerous physiological processes, including circadian rhythm, anxiety, memory,
fear, and stress, are regulated by NPY. NPY has anti-stress properties. In brain
areas linked to stress, it alters neurotransmission [74, 75]. By suppressing the
secretion of IFN-
Brain function depends on the coordinated activity of presynaptic and postsynaptic signals generated by neurons and non-neuronal components, such as glial cells. Inflammation via mediators, such as cytokines, affects neuronal differentiation and synapse transmission. Although there is evidence for many reasons why stress causes inflammation and subsequently impacts cognitive functions, we are still far from fully understanding the underlying molecular and cellular mechanisms. This review examines the possible mechanisms of stress-induced inflammation, focusing on the relationship between inflammation and cognitive functions. Understanding how stress regulates inflammation and cognitive functions may lead to identifying novel targets for human attention, learning, memory, cognition, and, eventually, treating psychiatric diseases.
ŞKŞ conceptualized and designed the study. ŞKŞ and EA collected, analyzed literatures. Both authors contributed to editorial changes in the manuscript. Both authors read and approved the final manuscript. Both authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.