
- Academic Editor
Alzheimer’s disease (AD) is characterized by cognitive decline and abnormal protein accumulation. Copper imbalance and pyroptosis play significant roles in the pathogenesis of AD. Recent studies have suggested that dysregulated copper homeostasis contributed to β-amyloid accumulation, which may activate the NOD-like receptor protein 3 (NLRP3)-related pyroptosis pathway, promoting neuronal damages and AD progression. Therefore, the present study aims to investigates whether copper facilitates AD through exacerbating β-amyloid (Aβ) induced activation of NLRP3/Caspase-1/Gasdermin D (GSDMD)-mediated neuronal cell pyroptosis.
Mouse hippocampal HT-22 cells were cultured with Aβ1-42 oligomer for 24 h as AD Model group. CuCl2 treatment was administered to the AD cell model, and cell survivability levels were detected by Cell Counting Kit-8 (CCK-8), TdT-mediated dUTP nick end labeling (TUNEL), and other relevant kits. Mitochondrial function was evaluated using Mitochondrial membrane potential dye JC-1 and transmission electron microscopy (TEM). After intervention with the NLRP3 inhibitor MCC950, activation of the NLRP3/Caspase-1/GSDMD pathway by copper ions (Cu2+) was confirmed via Western Blot. Thioredoxin T (ThT) fluorescence assay was performed to observe the aggregation effect of Aβ induced by Cu2+ overload.
CuCl2 treatment of the AD cell model resulted in up-regulation of the levels of Lactate Dehydrogenase (LDH), Interleukin-1β (IL-1β), and IL-18 expression, which indicated activation of pyroptosis. We observed a significant decrease in mitochondrial membrane potential, mitochondrial swelling, and loss of mitochondrial cristae by fluorescence microscopy and TEM. ThT fluorescence imaging showed that Cu2+ promoted Aβ aggregation and up-regulated NLRP3, apoptosis-associated speck-like protein containing a CARD (ACS), Caspase-1, Cleaved Caspase-1, GSDMD, and Gasdermin D N-terminal (GSDMD-NT). The NLRP3 inhibitor MCC950 partially reversed Cu2+-mediated pyroptosis in HT-22 cells.
Exposure to copper ions disrupt mitochondrial copper homeostasis, promotes Aβ aggregation, and activates NLRP3 inflammasomes, further promoting the Aβ aggregation activated pyroptosis in AD cell models.
Alzheimer’s disease (AD) is a common progressive neurodegenerative disease. The
clinical manifestations of AD include continuous cognitive and memory decline,
reduced ability to perform daily living activities, and a variety of
neuropsychiatric symptoms and behavioral disorders [1, 2]. Abnormal accumulation of
Accumulative studies have indicated the critical role of copper in various
cellular processes, particularly in mitochondrial function, metabolic
reprogramming, and cancer biology, indicating the vital function of
copper-mitochondrial homeostasis in various diseases [5, 6, 7, 8, 9, 10, 11]. Proteomics revealed
protein targets and pathways in AD that are significantly altered by
copper-induced and oxidative modifications, which are related to the mitochondria
dysfunction [6]. Recent studies focusing on the effects of metal ions on AD also
determined the abnormal accumulation of copper ions (Cu2+), indicating that
copper level might be one of the pathological factors of AD pathogenesis [7, 8].
Elevated levels of Cu2+ may result in the aggregation of A
Pyroptosis is a novel mode of cell death triggered by activated inflammatory
vesicles, of which the inflammatory vesicles are represented by the NOD-like
receptor (NLR) family [12]. A
Mouse hippocampal cell HT-22 (CL-039m,
American Type Culture Collection (ATCC), Manassas, VA, USA). Dulbecco’s modified Eagle’s medium (DMEM) and Trypsin (12491015 and
25200072, Gibco, Grand Island, NE, USA). PBS buffer, Ammonium persulfate
substitute, Radio-immunoprecipitation assay buffer (RIPA), Phenylmethylsulfonyl
fluoride (PMSF), Sodium dodecyl sulfate (SDS), Tetramethylethylenediamine
(TEMED), and TdT-mediated dUTP nick end labeling (TUNEL) assay kit (C0221A, ST005,
P0013B, ST505, ST626, ST728, and C1088, Beyotime, Shanghai, China). 30% Acr/Bic,
Tris-Base, Triton X-100, TBS buffer (BL513B, BS083-500g, BS084-1000ml, BL600A,
Biosharp, Hefei, China). SDS, Glycine, skimmed milk (3250KG001, 1275KG2P5, Biofroxx
GmbH, Guangzhou, China). Bicinchoninic acid (BCA) protein assay kit, 5
Ultra-low temperature refrigerator (DW-86L626, Haier, Qingdao, China). Optical microscope (XSP-2800, Shanghai Optical Instrument No.1 Factory, Shanghai, China). CO2 cell incubator, ultra-clean bench, constant temperature water bath (BC-J80, BSC-1000B2, SSW-420-2S, Shanghai BoXun Industrial Medical Equipment Factory, Shanghai, China). Ultra-pure water meter (ELGA Veoli, Paris, France). Real-time fluorescence quantitative polymerase chain reaction (PCR) instrument (CFX96 Touch 1855195, Bio-Rad, Hercules, CA, USA), Western blotting system Criterion™ electrophoresis tank+Trans-blot® transfer tank (1658033, Bio-Rad, Hercules, CA, USA). EVOS M5000 fluorescence microscope (AMF5000, Themo Fisher, Waltham, MA, USA). Orbital shaker TS-100 (E0020, Kylin-Bell, Haimen, China). JP-K6000 chemiluminescence analyzer (ZF-368, Shanghai Jiapeng Technology Co., Ltd., Shanghai, China).
The HT-22 cell line has been meticulously certified through analysis and has successfully passed routine mycoplasma contamination tests, with the outcome being negative. The HT-22 cells were cultured in DMEM supplemented with 10% FBS+1% P/S. When the cells reached 80%–90%, PBS was used for washing. Digestion was terminated by adding 2 mL of 0.25% trypsin-0.02% Ethylenediaminetetraacetic acid (EDTA, ST1303-250g, Beyotime, Shanghai, China) mixture and 6 mL of complete medium. Cells were resuspended after centrifugation for cell counting and plate spreading.
Routinely cultured HT-22 cells were used as controls. HT-22 cells were cultured
with 10 µM concentration of A
HT-22 cells were seeded into 24-well plates with cell crawlers at a density of 1
HT-22 cells were seeded into 24-well plates with cell inserts at a density of 1
HT-22 cells in the logarithmic growth phase were seeded into 96-well plates at a density of 2000 cells per well. Maximum enzyme activity control wells were added with LDH release reagent. 120 µL of supernatant from each well was taken and 60 µL of LDH assay working solution was added, and the absorbance was measured at 490 nm.
The supernatant of HT-22 cells in each group was collected, and the standard was prepared according to the instructions of the ELISA kit. The sample was added 50 µL and incubated at 37 °C for 30 min. After washing, the enzyme-labeled reagent was added and the absorbance of each hole was measured at 450 nm.
HT-22 cells were seeded at a density of 2000 cells per well into 96-well plates with 100 µL of medium. Cells were cultured in 5 % CO2, 37 °C for 24 h. 10 µL CCK-8 solution was added to each well and incubated for 2 hours. Absorbance was measured at 450 nm with an enzyme marker.
HT-22 cells were seeded at a density of 1
Total protein was extracted with RIPA lysate and protein quantification was performed by BCA method. Proteins were separated via SDS-PAGE gel electrophoresis and transferred to a Polyvinylidene fluoride (PVDF) membrane (ISEQ00010, Millipore, Burlington, MA, USA). The membranes were then closed with 5% skimmed milk for 2 h at room temperature and incubated with NLRP3, ASC, Caspase-1, Cleaved Caspase-1, GSDMD, Gasdermin D N-terminal (GSDMD-NT) primary antibody at 4 °C overnight and incubated by secondary antibody. The membranes were developed with a chemiluminescent imaging system and analyzed in grayscale with ImageJ Verson2 software (Bethesda, Rockville, MD, USA). In this experiment, 1.0 mm 10-well gel, 12% separation glue and 5% concentration glue were used. The electrophoresis conditions were 80 V 20 min and 120 V 45 min, respectively. The membrane transfer condition was 200 mA in ice water bath for 90 min. The original figures of Western Blot can be found in the Supplementary Materials.
Data were analyzed and plotted using Graphpad Prism 9 (Version 9.4.0) (GraphPad Software, San
Diego, CA, USA). AI was used to organize the combined graphs. All data were
expressed as means
To verify the effect of copper overload on the mitochondrial function in HT-22
cells, we first used JC-1 to detect mitochondrial membrane potential. As shown in
Fig. 1A,B, the mitochondrial membrane potential was significantly lower in the AD
model group compared to the Control group (p
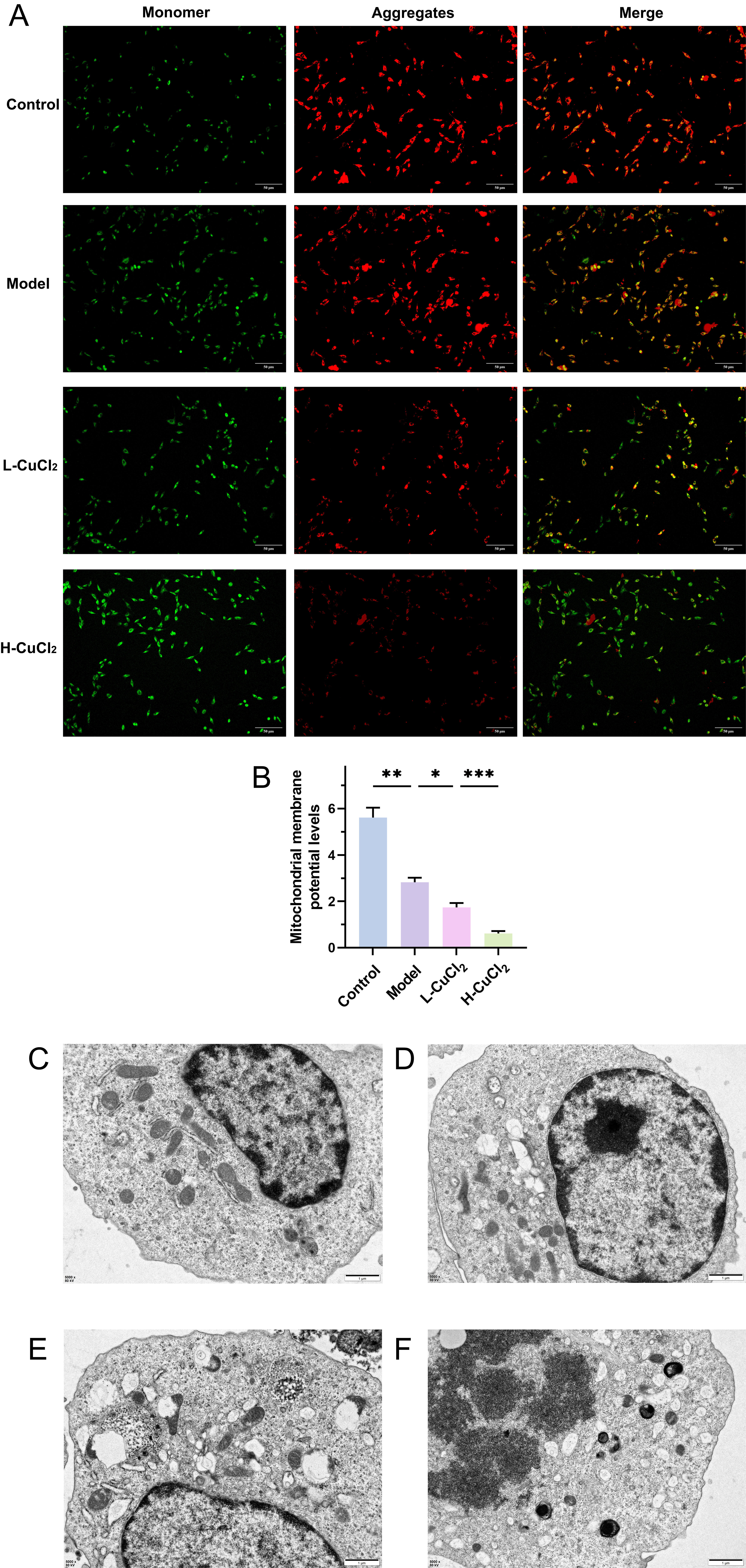 Fig. 1.
Fig. 1.
The mitochondrial membrane potential and cell pyroptosis
morphology in HT-22 cells were detected by JC-1 staining. (A) fluorescent
images, (B) histogram. Transmission electron microscopy (TEM) images of (C)
Control, (D) Model, (E) L-CuCl2, and (F) H-CuCl2. A: scale bar = 50 µm, C–F: scale bar = 1 µm. *p
To investigate the effects of copper overload on the AD model of HT-22 cells, we
detected the cell death, LDH, IL-1
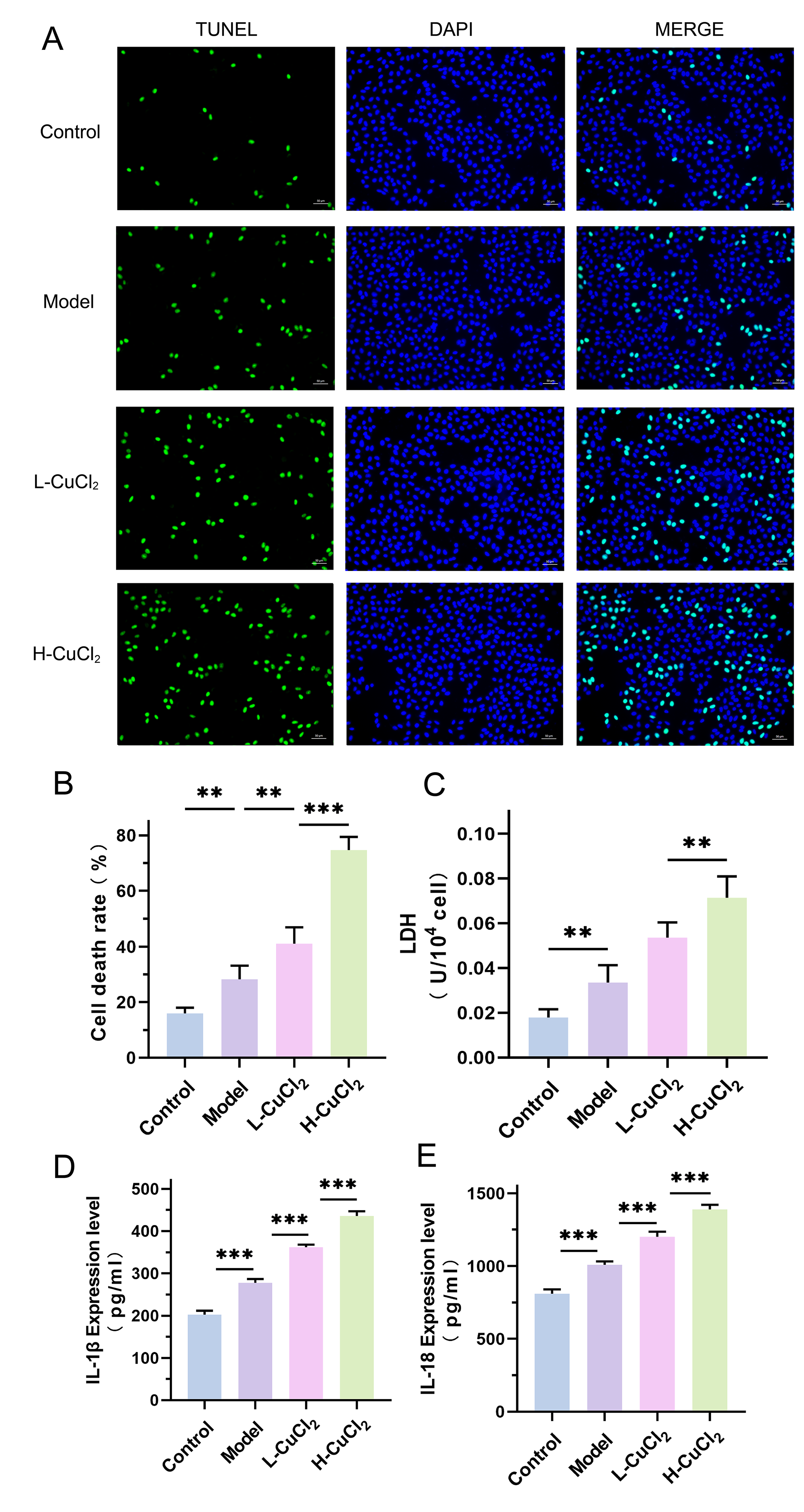 Fig. 2.
Fig. 2.
Cell death markers and inflammatory factors were detected. The
occurrence of cell death was detected by TUNEL staining (A) fluorescent images,
(B) histogram. (C) Lactate Dehydrogenase (LDH) levels were detected by LDH kit.
The levels of IL-1
To verify the activating cell death of Cu2+ was pyroptosis in HT-22 cells,
we detected pyroptosis pathway proteins by Western Blot. As shown in Fig. 3, Cleaved
Caspase-1 and GSDMD-NT expression was not detected in the Control group. However,
the expression levels of NLRP3, ASC, Cleaved Caspase-1, and GSDMD-NT were all
significantly in the AD model of HT-22 cells (p
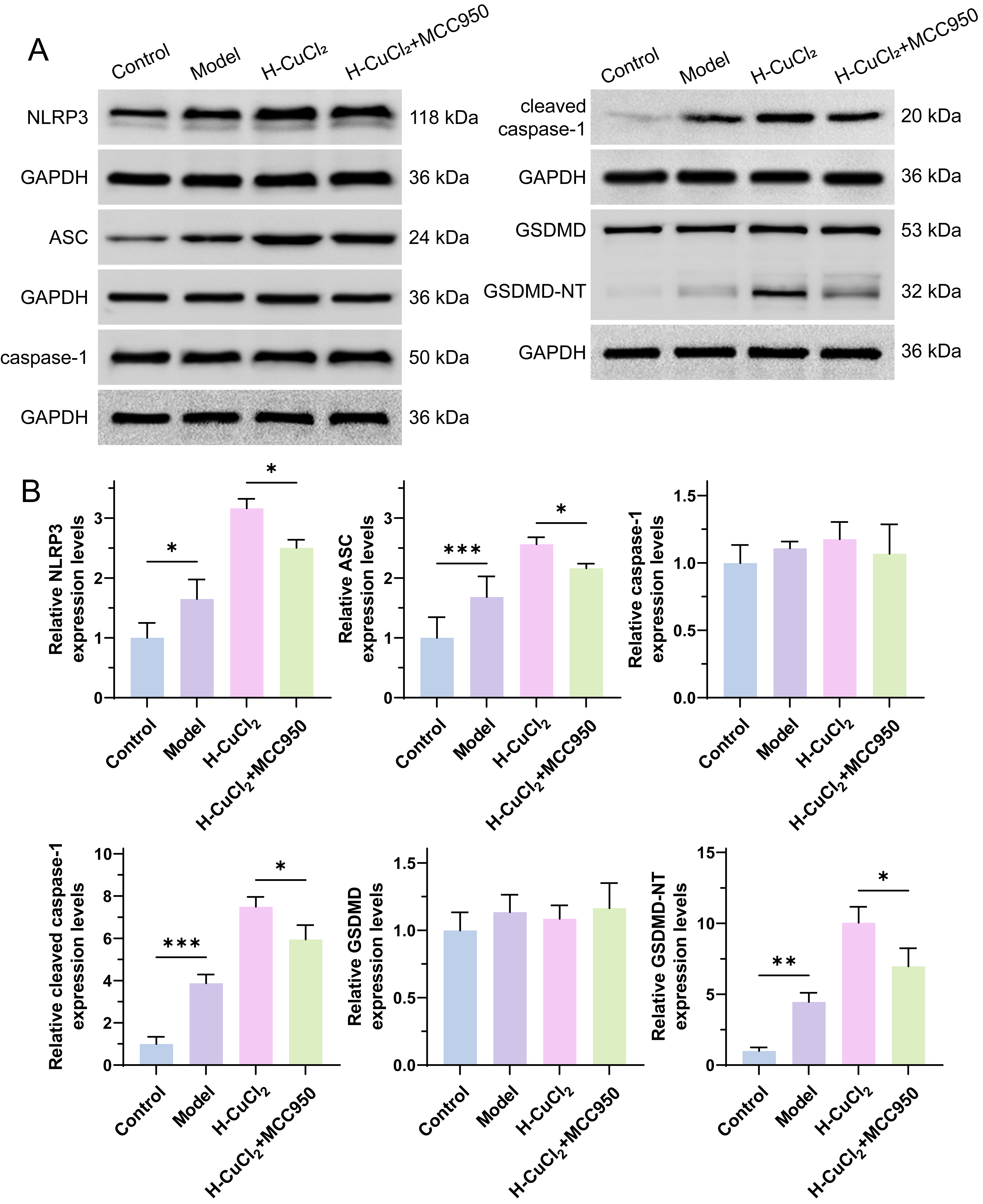 Fig. 3.
Fig. 3.
Pyroptosis related proteins were detected. (A) Western blot
assay for the protein expression levels of NLRP3, ASC, Caspase-1, Cleaved
Caspase-1, GSDMD, and GSDMD-NT. (B) Quantitative analysis of the protein
expression levels of NLRP3, ASC, Caspase-1, Cleaved Caspase-1, GSDMD, and
GSDMD-NT. *p
To investigate how copper overload aggravates pyroptosis in the AD model of
HT-22 cells, we observed A
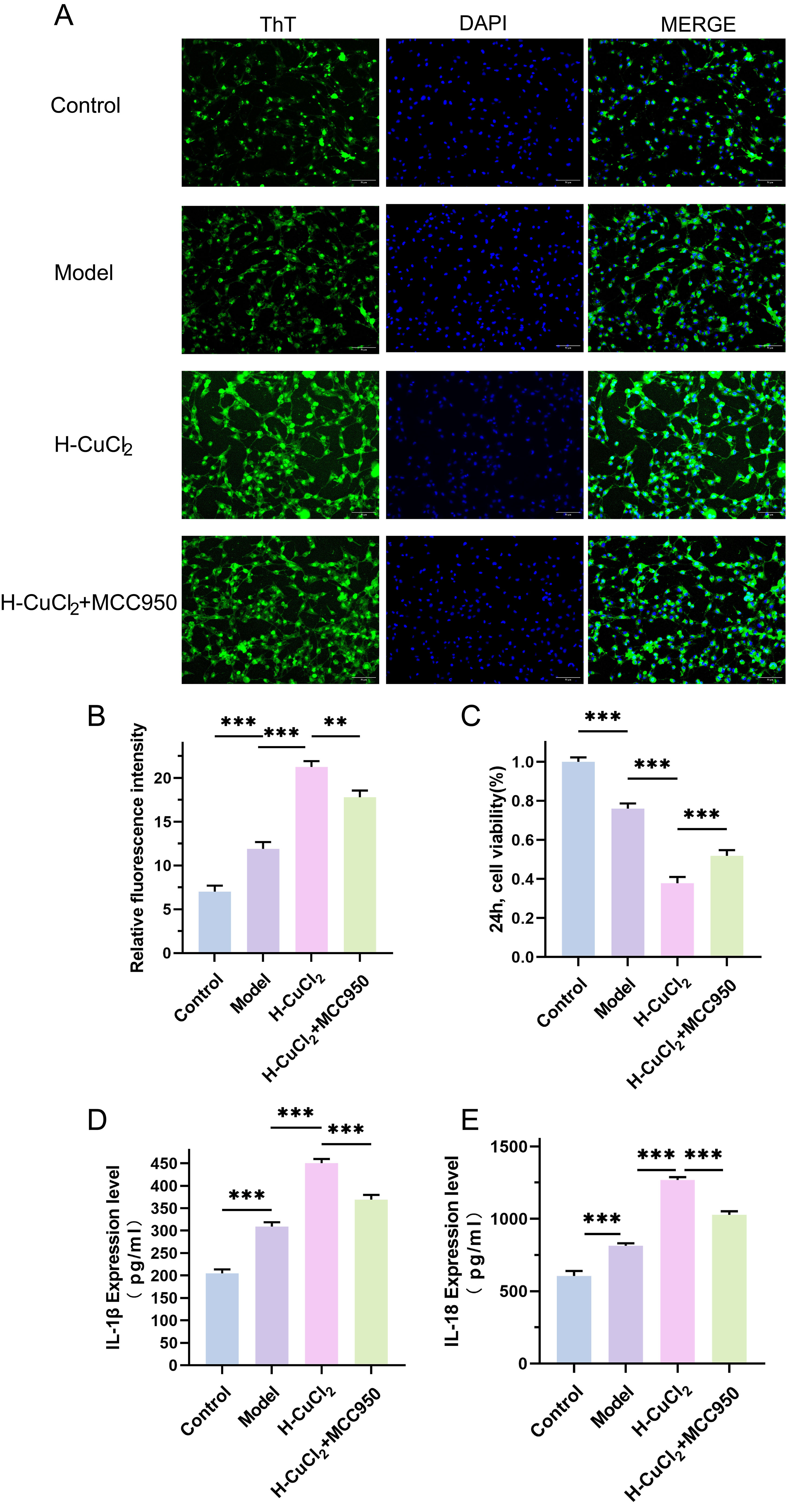 Fig. 4.
Fig. 4.
Thioflavin T (THT) fluorescence staining was used to observe the
aggregation of
Copper is an essential trace element in human physiological system [17]. The
brain is abundant in mitochondria, serving as a “copper reservoir”, which is
crucial for cellular activities; consequently, it boasts the highest copper
content among all organs [18]. When the intracellular copper concentration
surpasses the threshold maintained by the homeostatic mechanism, it exerts toxic
effects on the cells. This can directly impact neural development and energy
metabolism, ultimately leading to severe neurological damage [19]. Study has
indicated that serum copper levels in AD patients were approximately 54% higher
than those in healthy individuals, suggesting the potential value of copper
levels as a peripheral blood marker for oxidative stress in distinguishing AD
[20]. In the present study, we observed that Cu2+ treatment downregulated
mitochondrial membrane potential and exacerbated mitochondrial damage in the AD
model of HT-22 cells. At the same time, pyroptosis in HT-22 cells was observed by
TUNEL staining, and the expression levels of the pyroptosis markers LDH,
IL-1
A
In conclusion, we explored the effects of copper overload on
A
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Conceptualization: MJZ, CPW; Methodology: MJZ, LZ, CPW; Formal analysis and investigation: MJZ, LZ, CPW; Writing—original draft preparation: MJZ; Writing—review and editing: CPW; Funding acquisition: CPW; Supervision: CPW. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
The present study was supported by the Interdisciplinary Program of Shanghai Jiao Tong University (No. YG2021QN89) from Chang-Peng Wang.
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/j.jin2310194.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.