
- Academic Editors
Developmental language disorders (DLDs) are common neurodevelopmental conditions, affecting approximately 7–10% of children, with significant impacts on communication, academic achievement, and social integration. While genetic factors are known contributors, the underlying genomic architecture and biological pathways remain incompletely understood. This analysis explores key genomic biomarkers of DLD and investigates their functional interactions.
We conducted an integrative genomic analysis combining multiple data-driven approaches. Using the Open Targets platform, we compiled a set of genes associated with DLD-related phenotypes (based on evidence scores ≥0.3) and constructed a gene-phenotype network to visualize these associations. Protein-protein interaction mapping of the identified genes was performed using the STRING database to uncover interaction clusters and shared pathways. We then analyzed sequence and structural relationships among the encoded proteins, including pairwise sequence homology (BLAST alignments), 3D structural modeling, and multimeric interaction prediction using AlphaFold 3.
Our analysis identified 89 genes linked to 14 DLD-related phenotypic terms, with strong clustering around delayed speech. Several genes (e.g., GRN, MAPT, FOXP2, FOXP1, AP4E1) showed particularly high-confidence associations. Structural analysis of encoded proteins revealed unexpected similarity between functionally related but sequence-divergent pairs (e.g., WDR45 and GNB1). AlphaFold 3 modeling predicted a potential interaction between DCDC2 and KIAA0319, suggesting a plausible structural mechanism for their co-involvement in dyslexia.
DLDs emerge from diverse genetic contributors but converge on shared neurodevelopmental pathways. Structural modeling enhances genomic insights by uncovering hidden relationships and candidate interactions, paving the way for more precise genetic screening and functional studies in language disorders.
Language is a defining feature of human cognition, enabling the communication of complex ideas and the development of cultures [1]. Studies suggest that spoken languages evolved from ancient communication systems that relied on gestures and arm movements [2]. Supporting this theory, research on human infants demonstrates synchronization between gestures and vocalizations as early as 2 to 3 months old, with older infants increasingly integrating gestures with word comprehension and production [3, 4, 5].
While language development is a hallmark of typical human growth, some individuals experience language disorders—conditions that impair their ability to learn and communicate. These disorders manifest in various forms, including difficulties with speech production, language comprehension, or coherent verbal expression. Many children with language disorders are often misclassified as having learning disabilities or behavioral problems, further impeding their educational and social development [6].
Early diagnosis and intervention are critical, as untreated language disorders often persist into adulthood, increasing the risk of social exclusion, unemployment, and mental health conditions such as anxiety and depression [7]. Childhood language disorders have been linked to difficulties in forming peer relationships and heightened susceptibility to bullying, exacerbating social challenges [8, 9]. Research associates language disorders with structural and functional abnormalities in key brain regions, particularly in the left hemisphere, including Broca’s and Wernicke’s areas [10]. Environmental factors, such as socioeconomic status and early language exposure, may influence the severity and outcomes of language disorders [11].
Genomic studies have identified key genes such as FOXP2, CNTNAP2, ATP2C2, and CMIP that contribute to the development of speech and language, reinforcing the biological foundation of language evolution [12, 13]. This genomic perspective has advanced the precision of diagnoses and personalized therapies by pinpointing genetic markers, thereby improving our understanding of the acquisition of language disorders and the neuropathology of associated developmental disorders [14].
This review provides a comprehensive perspective on genomic studies, exploring genetic markers and their associations with various language impairments. It employs network analysis to examine gene-phenotype relationships in developmental language disorders (DLDs) and investigates sequence and structural similarities among biomarkers to uncover shared biological roles. Additionally, the study addresses challenges in researching language disorders and offers recommendations for future research directions.
Unlike acquired language disorders, which result from stroke or traumatic brain injury, such as aphasia, developmental language disorders (DLDs) typically emerge during early childhood, affecting approximately 6–8% of the population [15]. These disorders persist into adolescence and adulthood if not addressed through timely and appropriate interventions. Unlike acquired language disorders, DLDs occur in the absence of neurological damage, hearing impairment, or global cognitive deficits [16]. Instead, they are characterized by persistent difficulties in acquiring and using language across various modalities, including speaking, reading, and writing [17].
Historically, the terms Developmental Language Disorder (DLD) and Specific Language Impairment (SLI) have been used to describe children with persistent language difficulties, though their definitions and clinical usage have evolved over time. Specific Language Impairment (SLI) was traditionally used to describe language difficulties in children with normal cognitive abilities and no clear underlying cause [18]. It affects approximately 7–10% of kindergarten-aged children [19] and manifests through challenges in speaking, listening, reading, and writing [19]. The term Developmental Language Disorder (DLD) was introduced through the CATALISE Consortium [17] to replace SLI and provide a more inclusive and clinically useful framework. Unlike SLI, DLD does not require strict exclusion criteria based on cognitive abilities or co-occurring conditions, making it a more comprehensive diagnosis.
DLDs can manifest in several domains:
• Speech sound disorder: Characterized by persistent difficulties in producing speech sounds, which can affect communication clarity and literacy development [20]. It has been associated with abnormalities in Broca’s area, a key brain region involved in articulation and speech production [21].
• Stuttering: Involves speech disruptions, such as repeated sounds, pauses, or prolonged syllables, often accompanied by involuntary movements [22]. Neuroimaging studies have linked stuttering to dysfunctions in Broca’s area [23], the left superior temporal gyrus [24], the cortico-basal ganglia-thalamocortical loop [25].
• Receptive language difficulties: These involve reading comprehension problems [26].
• Reading disorders: Many children with DLDs exhibit dyslexia or other reading impairments, characterized by difficulties in phonological processing, word recognition, and reading fluency [27]. Neuroimaging studies reveal atypical activation patterns in left-hemisphere language regions, including right parietal lobe dysfunction [28] and increased activation in the left inferior frontal gyrus in the frontal lobe during reading tasks [29].
• Writing difficulties: Impairments in spelling, sentence structure, and coherence are often linked to underlying phonological and grammatical deficits [30].
The genomics of language remains an open and complex question. Language is a uniquely human trait, yet its genetic basis and origins remain unconfirmed. Over the past few decades, with the advances in molecular research, FOXP2 has emerged as a dominant focus in studies on language evolution and disorders.
In 2002, Enard et al. [1] examined the amino acid sequence of FOXP2 in humans and compared it with that of other species (e.g., chimpanzees, gorillas, orangutans, rhesus macaques, and mice). The study identified two amino acid substitutions in the human FOXP2 sequence (T303N and N325S) out of 715 amino acids relative to the other species, as well as one additional substitution when compared to mice. The authors speculated that these mutations may have conferred an evolutionary advantage, potentially linked to the emergence of language in humans.
However, this hypothesis has been debated. For instance, one study argued that these FOXP2 mutations arose before the emergence of modern humans [31], challenging the direct link between FOXP2 mutations and language evolution. As a result, the question of language origins remains unresolved and subject to further investigation. It has not been possible to accurately determine the structural role of the two residues (T303 and N325) due to the absence of a full-length, experimentally determined FOXP2 structure. The only available structure in the Protein Data Bank (PDB: 2AS5) includes just the DNA-binding domain—approximately 100 residues out of the 715 in the full-length protein. With the advent of AlphaFold3 [32], the most accurate protein structure prediction algorithm to date, we were able to predict the full-length FOXP2 structure and map the locations of these two residues. As shown in Fig. 1, and consistent with earlier hypotheses, T303 and N325 (highlighted in red) are located in an unstructured (disordered) region N-terminal to the zinc-finger domain and lie outside both the DNA-binding forkhead domain (FHD) and the zinc-finger/leucine-zipper regions—domains critical for FOXP2’s transcriptional regulatory function. This suggests that these two variants are unlikely to contribute to human-specific selection. Further insights into the disordered regions of FOXP2 were recently provided by Adnan et al. [33], who suggested that these regions may become ordered upon hexamerization.
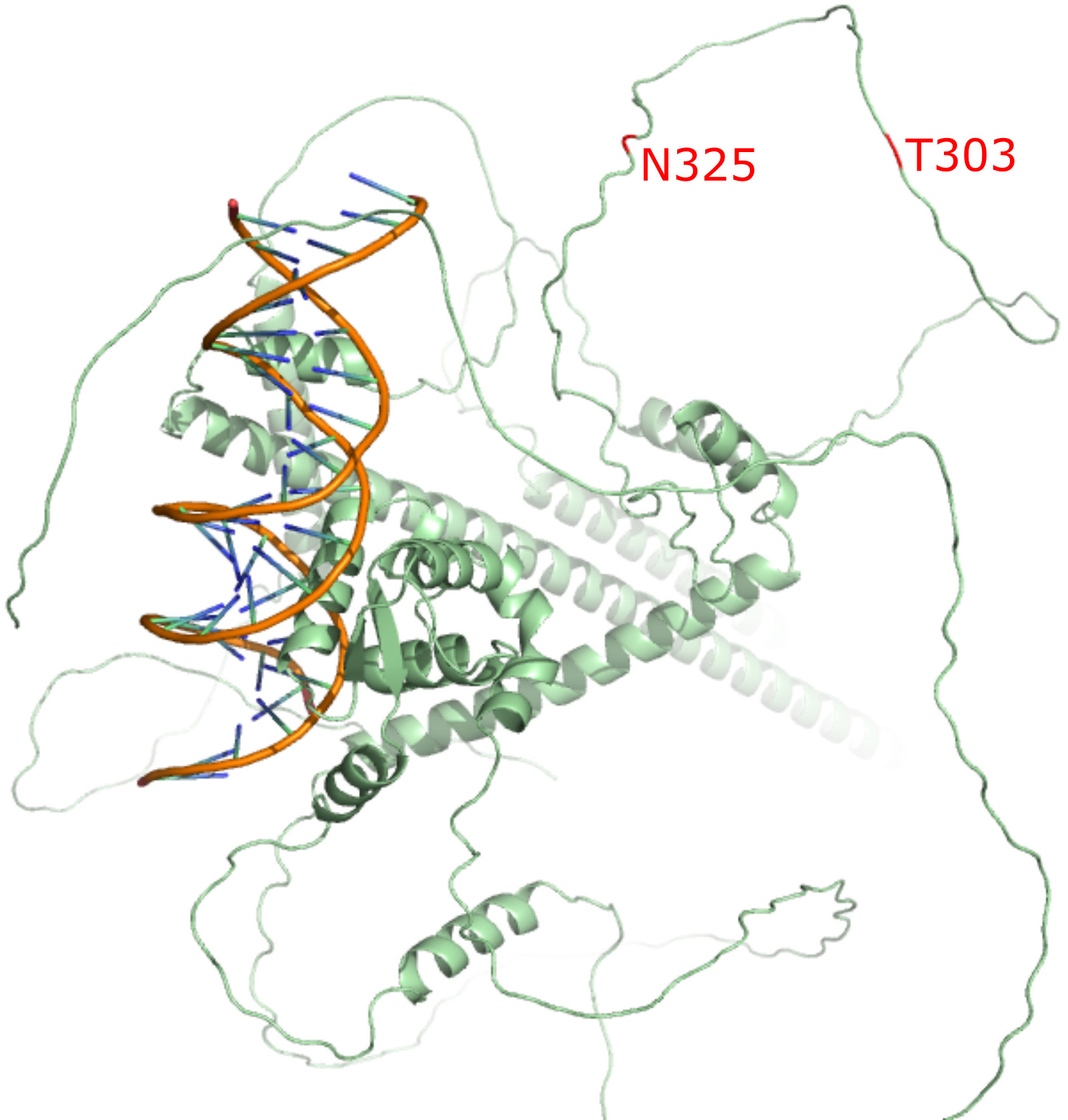 Fig. 1.
Fig. 1.
The predicted 3D structure of FOXP2 (AlphaFold 3) superimposed on the DNA-binding domain from PDB (PDB: 2AS5). The T303N and N325S residues are highlighted in red.
While the relationship between the two FOXP2 mutations and evolution remains questionable and debated, this does not dispute the crucial role of FOXP2 in vocalization. Even without recent selection in humans, the unusual effects of FOXP2 dysfunction mean that it is still a promising window into the neurobiology of speech and language [34]. FOXP2 is one of the most extensively studied genes in relation to language disorders and was the first gene to be associated predominantly with speech and language disorders [35]. Experiments on mouse models have highlighted its role in developmental processes related to social communication functions [36]. Except for the mentioned residues, the FOXP2 protein in humans and mice is identical, including its expression pattern, making the mouse model a suitable system for studying its function [35]. It has been found that the disruption of a single copy of the FOXP2 gene in mice leads to modest developmental delays and significant alterations in ultrasonic vocalization. In contrast, the disruption of both copies results in severe motor impairments, premature death, and a complete absence of ultrasonic vocalizations in response to stressors [35]. Further support for forkhead box protein P2 (FOXP2)’s role in vocal communication comes from studies in other species. In canaries, Foxp2 expression in Area X—a brain region involved in song learning—fluctuates seasonally, with higher expression levels observed when songs are more variable. This pattern mirrors the role of FOXP2 in human speech plasticity [35]. Additionally, studies in fruit flies have revealed that the dFoxP2 gene is essential for operant self-learning, a cognitive process that shares similarities with aspects of language acquisition [37]. Many nonsense and missense mutations in FOXP2 have been reported as pathogenic in ClinVar [38], primarily associated with childhood apraxia of speech. These include variants such as R328*, R375*, R381, R477*, R553H, R578H, R552H, R570H, and R563*.
While FOXP2 has been a focal point of research, it represents only one part of the complex genetic landscape of language, which involves other genes [39]. Broader genomic studies suggest that DLDs have a complex polygenic basis [40], which involves multiple genes. Furthermore, some studies have failed to replicate FOXP2 associations with DLDs, highlighting the need for larger, well-powered cohorts and multi-gene analyses. For example, the GRIN2B gene contributes to neurogenesis and cognitive processes [41], including short-term memory and language acquisition [42], and is associated with reading difficulties, such as dyslexia [43].
Advances in genomics have led to the identification of key genes that could serve as potential biomarkers for language disorders, including CNTNAP2, DOCK4, GTF2I, SLC2A3, ATP2C2, CMIP, PCNT, DIP2A, S100B, and PRMT2 [43]. Other genes, such as KIAA0319 [44, 45, 46] and DCDC2 [47, 48, 49, 50] are strongly associated with reading disorders (e.g., dyslexia). Furthermore, variations in FOXP2 and ATP2C2 are more closely linked to speech and language impairments. These biomarkers play critical roles in neuronal connectivity, migration, and synaptic function, underscoring the complex genetic underpinnings of language impairments and hold promise for early diagnosis and personalized intervention.
A recent study [51] sequenced the whole genome of 19 individuals diagnosed with childhood apraxia and identified mutations in a set of genes associated with speech proficiency (CHD3, SETD1A, WDR5, KAT6A, SETBP1, ZFHX4, TNRC6B, and MKL2). Their findings revealed gene regulatory pathways in the developing brain that may contribute to the acquisition of proficient speech.
Language disorders exhibit considerable heterogeneity in both their presentation and genetic basis. While some genes are associated with specific language impairments, others contribute to broader neurodevelopmental pathways that overlap with conditions such as Autism Spectrum Disorder (ASD) and Attention Deficit Hyperactivity Disorder (ADHD). This complexity highlights the need to distinguish between shared genetic factors that contribute to multiple disorders and distinct genetic markers unique to specific language impairments.
Recent large-scale genomic studies have begun to shed new light on the complex genetic architecture of language-related traits and developmental language disorders. A landmark study by the GenLang Consortium conducted genome-wide association meta-analyses across five reading- and language-related traits in over 30,000 individuals, identifying heritable components and shared genetic factors with cognitive and neuroanatomical traits [52]. Similarly, a recent large-scale Danish study of DLD genetics used genotype and language-related data from over 25,000 individuals to estimate SNP-based heritability for DLD, reporting values between 27% and 52%, and highlighting the role of common genetic variants in language difficulties [53]. These studies emphasize the polygenic and multifactorial nature of DLD, aligning with the direction and motivation of our current investigation.
We obtained our datasets from the Open Targets Platform (version 24.03) [54] and extracted biomarker–disease association scores by integrating evidence from diverse sources, including genetic studies, somatic mutation datasets, known drug interactions, RNA expression data, and computational predictions.
To ensure the relevance of genetic associations, we limited our search to phenotypes classified under the International Classification of Diseases, 11th Revision (ICD-11) for Mortality and Morbidity Statistics [55] (accessed on 2024-11-02), using the following diagnostic terms:
• 6A01 Developmental Language Disorders.
• 6A03 Developmental Learning Disorder.
• MA81 Speech Dysfluency.
• VV2Y Other Specified Voice and Speech Functions.
Other neurodevelopmental conditions, such as Autism Spectrum Disorder (ASD) and Intellectual Developmental Disorder (IDD), were excluded from the primary analysis to maintain specificity. However, they are discussed in the main text when biomarkers showed significant associations with these conditions.
To refine the dataset, we applied an additional filter, retaining only biomarker–disease associations with an Open Targets score above 0.3. This threshold ensured that only associations supported by stronger or more consistent evidence were included in the final analysis. These filtered associations were subsequently used in the network analysis, and the corresponding biomarkers were reported.
For further discussion and contextualization, the identified biomarkers were also investigated in PubMed to retrieve relevant peer-reviewed publications supporting their reported associations with developmental language disorders.
We evaluated sequence and structural similarities for proteins encoded by the selected biomarkers using pairwise comparisons. Protein sequences were aligned using BLAST 2.8.1+, and sequence identity scores were calculated as the product of alignment coverage and percent identity. Structural similarity was assessed using the TM-score, calculated with USAlign [56]. Protein structures were visualized with PyMol 2.5.0. Predicted 3D structures were retrieved from the EBI AlphaFold Protein Structure Database [57] on 2024-09-02.
Protein structure models for individual biomarkers were obtained from the AlphaFold Protein Structure Database hosted by EMBL-EBI (https://www.alphafold.ebi.ac.uk/), which provides high-confidence predictions of monomeric protein structures. For the analysis of potential protein–protein interactions between KIAA0319 and DCDC2, we utilized AlphaFold 3, a recently released model capable of multimeric complex prediction. The multimeric structure prediction was used to examine possible binding interfaces and assess whether known genetic variants coincide with residues at the predicted interaction site.
Network analysis was performed to identify clusters of closely related disorders based on shared genetic associations. Using association scores from the Open Targets dataset, we constructed a network in which nodes represent either disorders or genes, and edges denote the strength of associations. The network was generated using Python 3 libraries: networkx, scipy, and sklearn. Clusters were identified using a community detection algorithm to highlight genetic pathways commonly implicated across language-related disorders. Cluster significance was assessed by measuring inter-cluster distances and evaluating association strength thresholds. Descriptive statistics and visualizations were generated to facilitate comparison across disorder categories.
We analyzed the protein–protein interaction (PPI) network of the identified genes using STRING-db (Search Tool for the Retrieval of Interacting Genes/Proteins) [58]. To enhance interpretability, nodes were color-coded based on phenotype classifications from The Monarch Initiative [59], a comprehensive resource for integrating genotype–phenotype relationships.
We obtained our dataset from the Open Targets platform [54], focusing specifically on phenotypes related to developmental language disorders (DLDs), as defined by the International Classification of Diseases, 11th Revision (ICD-11) for Mortality and Morbidity Statistics [55].
Using the Open Targets platform’s integrative evidence scoring system (range: 0 to 1), we assembled an updated dataset comprising 89 genes associated with 14 DLD-related phenotypes, each with an association score of
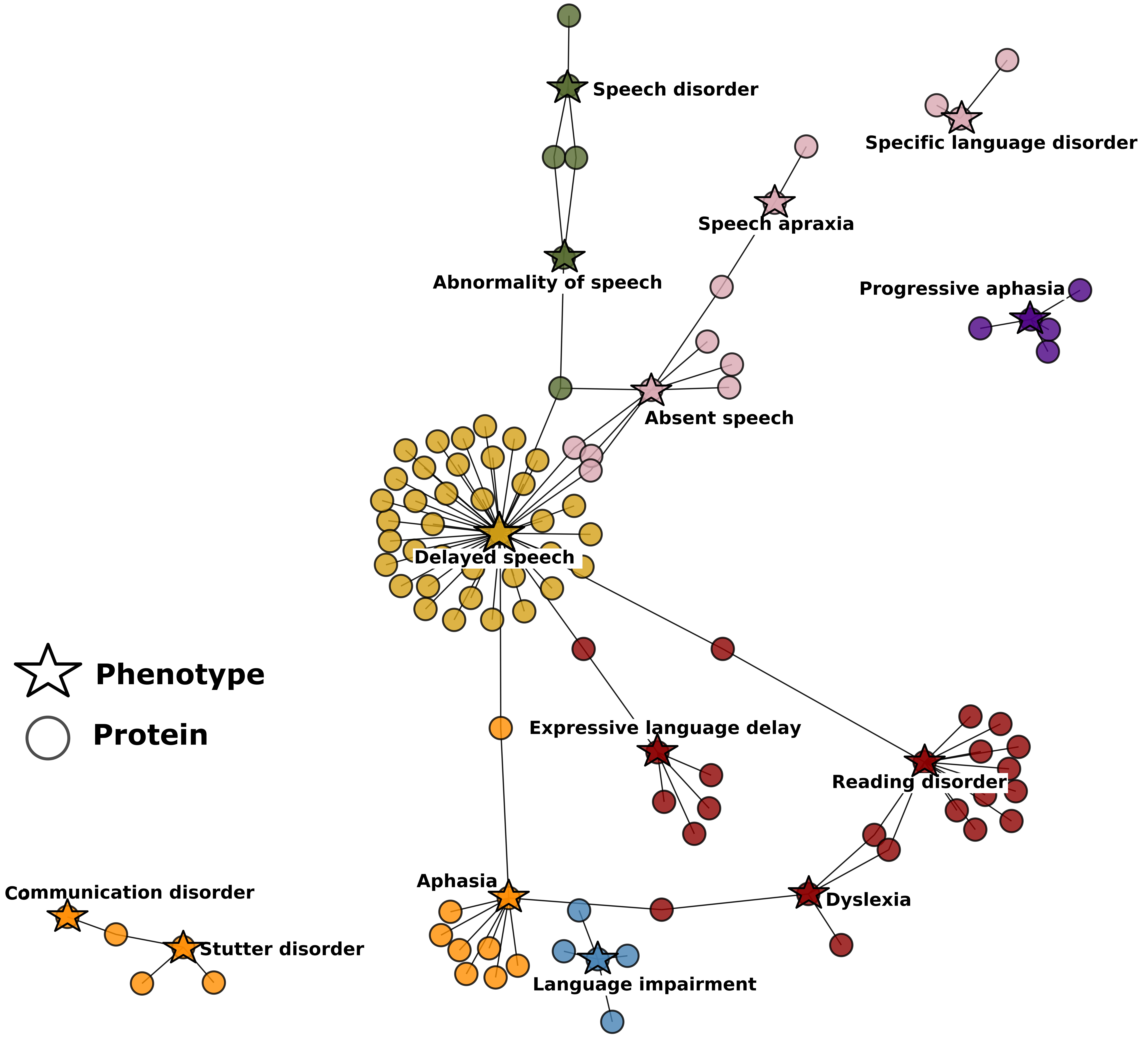 Fig. 2.
Fig. 2.
Network representation of genetic associations in developmental language disorders. Nodes represent genes or phenotypes, while edges denote association scores, highlighting clusters of related biomarkers and disorders.
| Gene | Associated phenotype | Association score |
| GRN | Progressive aphasia | 0.78 |
| MAPT | Progressive aphasia | 0.77 |
| FOXP2 | Specific language disorder | 0.77 |
| FOXP1 | Language impairment | 0.73 |
| AP4E1 | Stutter disorder | 0.72 |
| PSEN1 | Progressive aphasia | 0.70 |
| DNAAF4 | Dyslexia | 0.61 |
| TARDBP | Progressive aphasia | 0.56 |
| ARFGEF1 | Delayed speech | 0.53 |
| PURA | Delayed speech | 0.52 |
| AHDC1 | Delayed speech | 0.51 |
| BPTF | Delayed speech and Expressive language delay | 0.51 |
| ARID1B | Abnormality of speech, delayed speech and absent speech | 0.50 |
| GRIN2A | Aphasia | 0.46 |
| MECP2 | Delayed speech and absent speech | 0.46 |
| PRR12 | Delayed speech | 0.46 |
| GRIN2C | Aphasia | 0.46 |
| TMEM222 | Delayed speech | 0.46 |
| PPP2CA | Language impairment | 0.46 |
| GRIN3A | Aphasia | 0.46 |
| GRIN3B | Aphasia | 0.46 |
| GRIN2D | Aphasia | 0.46 |
| GRIN1 | Aphasia | 0.46 |
| GRIN2B | Aphasia and delayed speech | 0.46 |
| UFSP2 | Delayed speech and absent speech | 0.46 |
| PABPC1 | Expressive language delay | 0.46 |
| DCDC2 | Dyslexia | 0.46 |
| GNB1 | Expressive language delay | 0.45 |
| ACHE | Aphasia | 0.43 |
| EBF3 | Expressive language delay | 0.43 |
| NPC1 | Speech apraxia | 0.43 |
| WDR45 | Delayed speech and absent speech | 0.43 |
| TM4SF20 | Language impairment | 0.41 |
| DRD3 | Aphasia and Dyslexia | 0.39 |
| KIAA0319 | Dyslexia | 0.39 |
| H3-3B | Delayed speech | 0.39 |
| H3-3A | Delayed speech | 0.39 |
| GABBR1 | Delayed speech | 0.38 |
| SYNGAP1 | Delayed speech | 0.38 |
| SCN3A | Reading disorder | 0.37 |
| SCN1A | Reading disorder and Delayed speech | 0.37 |
| NF1 | Delayed speech | 0.37 |
| SCN2A | Reading disorder | 0.37 |
| SCN4A | Reading disorder | 0.37 |
| ASXL1 | Delayed speech | 0.37 |
| POU3F3 | Delayed speech | 0.37 |
| SCN11A | Reading disorder | 0.37 |
| ZMYND8 | Delayed speech | 0.37 |
| SCN5A | Reading disorder | 0.37 |
| SCN10A | Reading disorder | 0.37 |
| SCN8A | Reading disorder | 0.37 |
| LINGO4 | Speech disorder | 0.37 |
| CTR9 | Delayed speech | 0.37 |
| MED13 | Delayed speech | 0.37 |
| SCN9A | Reading disorder | 0.37 |
| BCL11A | Delayed speech | 0.34 |
| MED13L | Delayed speech | 0.34 |
| STAT1 | Delayed speech | 0.34 |
| TMCO1 | Delayed speech | 0.34 |
| CDKL5 | Delayed speech | 0.34 |
| STXBP1 | Delayed speech | 0.34 |
| POGZ | Speech apraxia and absent speech | 0.34 |
| OTUD7A | Language impairment | 0.34 |
| RPS6KA3 | Delayed speech | 0.34 |
| CLCN6 | Speech disorder and abnormality of speech | 0.34 |
| DHX30 | Delayed speech | 0.34 |
| DDX3X | Delayed speech | 0.34 |
| SCAPER | Abnormality of speech | 0.34 |
| HDAC8 | Delayed speech | 0.34 |
| SHANK1 | Delayed speech | 0.34 |
| PCDH19 | Delayed speech | 0.34 |
| TMEM147 | Absent speech | 0.34 |
| MTHFR | Delayed speech | 0.33 |
| TMEM67 | Absent speech | 0.33 |
| PQBP1 | Delayed speech | 0.33 |
| GSPT2 | Delayed speech | 0.33 |
| RAB3GAP1 | Absent speech | 0.33 |
| UBE3A | Expressive language delay | 0.33 |
| TANGO2 | Delayed speech | 0.33 |
| ANKRD11 | Delayed speech | 0.33 |
| NSD1 | Delayed speech | 0.33 |
| COPB1 | Delayed speech | 0.33 |
| TMC1 | Delayed speech | 0.33 |
| DRD2 | Stutter disorder and communication disorder | 0.33 |
| NR3C1 | Specific language disorder | 0.32 |
| MCOLN1 | Delayed speech | 0.32 |
| HTR2A | Stutter disorder | 0.32 |
| ACTL6A | Delayed speech | 0.31 |
| NKAIN3 | Reading disorder | 0.31 |
The network reveals a highly connected cluster centered around the “delayed speech” phenotype, which is associated with the largest number of genes—reflecting its broad definition and high prevalence across diverse language disorders. “Stuttering” and “speech communication” tend to cluster together and appear more distant from the other phenotypes. In contrast, “speech apraxia”, “absence of speech”, “abnormality of speech” and related speech disorders cluster closely together, reflecting their phenotypic similarity and indicating shared genetic underpinnings among these DLD subtypes. Phenotypes such as “Reading disorder”, “Dyslexia”, and “Progressive aphasia” appear more peripheral in the network, suggesting more specific or partially distinct genetic associations.
Six genes in our study achieved especially high association scores (
GRN mutations are a known genetic cause of primary progressive aphasia (PPA), frequently presenting with language disorders such as impaired grammar, reduced fluency, and word retrieval difficulties [60]. Many affected individuals fall into the nonfluent variant PPA otherwise specified, the latter marked by prominent linguistic deficits with relatively preserved articulation [60]. These cases often exhibit asymmetrical cortical atrophy involving the insula and dorsolateral prefrontal cortex. GRN mutations also contribute to tau-negative frontotemporal lobar degeneration, where speech abnormalities are common. Reduced progranulin levels in biofluids serve as a reliable biomarker for identifying mutation carriers and distinguishing them from sporadic cases [61].
MAPT mutations are closely linked to the nonfluent/agrammatic variant of PPA, characterized by nonfluent speech, agrammatism, and motor speech deficits such as apraxia and dysarthria [62, 63]. These language impairments correlate with left-lateralized tau pathology, especially in the mid-frontal cortex [62]. Acoustic markers such as reduced prosodic range and increased pause rate further reflect this phenotype and are associated with tau pathology and frontal atrophy [63]. While less frequent than GRN mutations, MAPT mutations define a distinct tau-related language disorder within the PPA spectrum.
FOXP2’s involvement in speech apraxia is well documented (as discussed in the Introduction), and its high association score here reinforces its centrality in speech and language pathology. FOXP1, a closely related gene encoding a transcription factor important for early development across multiple organ systems, is also highly ranked. Mutations or deletions in FOXP1 can disrupt its function and cause neurodevelopmental disorders [64]. The inclusion of FOXP1 among the top hits is consistent with its known phenotype (“FOXP1 syndrome”) of severe language impairment and confirms that both FOXP paralogs are among the strongest genetic contributors to DLD. Mutations in FOXP1 are associated with intellectual disability, speech and language impairments, autistic features, and other neurodevelopmental issues [64, 65, 66, 67, 68].
Mutations in AP4E1 have been linked to intellectual developmental disorder with language impairment and stuttering [69, 70, 71, 72]. This association is also documented in the Eurofins Biomnis genetics test guide [73].
In our gene–phenotype network, we identified a subset of genes that were linked to more than one DLD-related phenotype, highlighting pleiotropic genetic effects across different language disorders. Several genes in our analysis appeared in multiple phenotype groupings. This pattern suggests that these genes impact fundamental neurodevelopmental processes influencing a range of language and cognitive outcomes.
ARID1B encodes a subunit of the BAF (SWI/SNF) chromatin-remodeling complex and is a critical regulator of neurodevelopment. Pathogenic variants in ARID1B are known to cause Coffin–Siris syndrome and related neurodevelopmental disorders [74], which are characterized by intellectual disability and speech-language impairments. In our analysis ARID1B is linked to three phenotypes: Abnormality of speech, Delayed speech, and Absent speech. This association is supported by multiple pathogenic or likely pathogenic variants, including rs879253746, rs879253747, rs879253745, rs879253856, and rs1057518918, as reported in ClinVar. Individuals with ARID1B-related disorders often exhibit disproportionate speech impairment relative to motor deficits, highlighting the gene’s specific role in language processing [75]. Other studies link ARID1B mutations to intellectual disability [76] and autism spectrum disorder [77].
Another prominent gene in our dataset was POGZ, which showed a notable association with DLD phenotypes. POGZ encodes a pogo transposable element-derived zinc finger protein involved in chromatin modulation and gene regulation in neurons [78]. De novo loss-of-function mutations in POGZ cause White–Sutton syndrome, a neurodevelopm ental disorder characterized by intellectual disability, autism spectrum traits, and striking speech and language deficits [79].
In our network, POGZ is associated with both Absent speech and Speech apraxia, mediated by distinct pathogenic variants: rs1553212868 and rs796052217, respectively. This gene encodes a chromatin regulator implicated in synaptic plasticity and neurodevelopment, providing a mechanistic basis for its role in complex speech impairments. From a translational perspective, POGZ is already included in many diagnostic gene panels for intellectual disability and autism.
Two X-linked genes, MECP2 and WDR45, were also highlighted in our results, each with well-established links to severe neurodevelopmental syndromes in which language impairment is a core feature. MECP2 is the gene mutated in Rett syndrome, an X-linked dominant disorder [80] that predominantly affects girls and is characterized by typical early development followed by regression of language and motor skills in infancy. Our analysis found MECP2 associated with DLD phenotypes, reflecting the profound impact of MECP2 mutations on language abilities. In classic Rett syndrome, virtually all patients experience loss of spoken language—previously acquired words are lost, and purposeful speech does not develop in most. In our network, both MECP2 and WDR45 show associations with Delayed speech and Absent speech. For MECP2, the variant rs61749715 is linked to Absent speech, while rs61752992 and rs267608463 are associated with Delayed speech. In WDR45, the pathogenic variant rs387907329 underlies both phenotypes, consistent with its role in BPAN (Beta-propeller Protein-Associated Neurodegeneration), a disorder involving early speech regression and neurodegeneration.
In contrast to the above genes, which often produce broad syndromic effects, the genes DCDC2 and KIAA0319 were associated in our analysis specifically with phenotypes related to reading disorders, aligning with their known roles in developmental dyslexia. Both DCDC2 and KIAA0319 have long been recognized as key susceptibility genes for dyslexia, a language-based learning disorder that affects reading acquisition [44, 45, 46, 47, 48, 49, 81].
Our network also shows an association involving the DRD3 gene (dopamine receptor D3), pointing to a role for neuromodulatory pathways in speech and language disorders. DRD3 is primarily known for encoding a dopamine receptor implicated in movement control and neuropsychiatric conditions [82] (such as schizophrenia and addictive behaviors). DRD3, a dopamine receptor gene, is potentially linked to both Dyslexia and Aphasia, based on suggestive association data. Moreover, DRD3 is a pharmacological target in Phase II clinical trials investigating Levodopa for the treatment of aphasia, underscoring its therapeutic relevance.
To validate the biomarker–disease associations identified through Open Targets, we performed a comprehensive protein–protein interaction (PPI) analysis using the STRING database (Search Tool for the Retrieval of Interacting Genes/Proteins). STRING integrates evidence from diverse sources—including experimental protein interaction data, text mining of literature, and computational predictions—to construct biologically meaningful interaction networks. Mapping the set of 89 candidate biomarker genes into STRING allowed us to evaluate how these proteins are functionally interconnected and to identify any additional interaction clusters among them.
The resulting STRING-derived PPI network (Fig. 3) revealed several dense, distinct clusters of interacting proteins, each cluster corresponding to specific biological processes. Most of the clusters are related to language and neurodevelopment. To enhance interpretability, we applied a phenotype-based color-coding scheme to the network nodes using classifications from the Monarch Initiative (a database integrating genotype–phenotype relationships). This scheme highlights key categories of developmental language disorder phenotypes within the network, with each phenotype category assigned a unique node color in (Fig. 3).
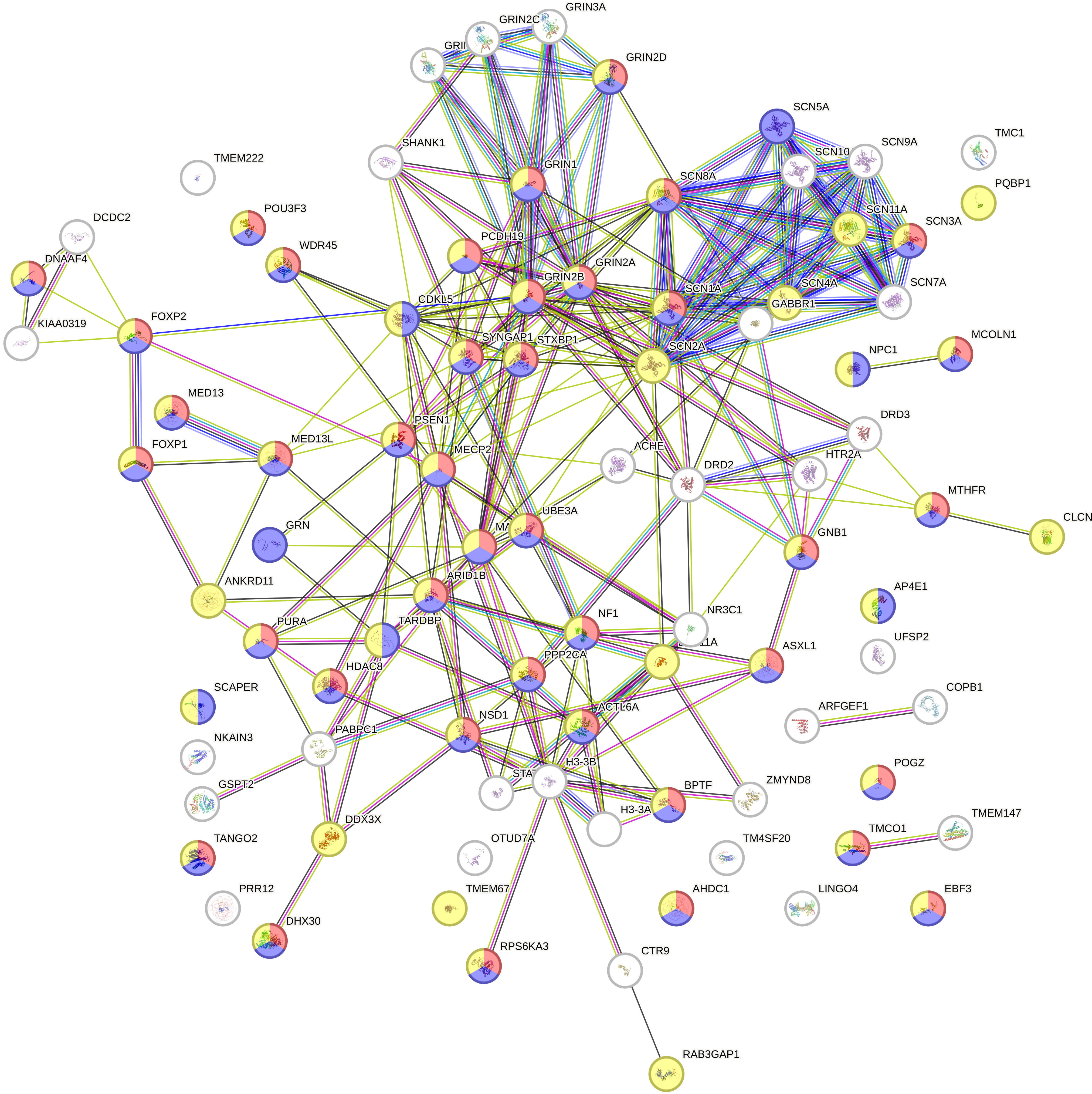 Fig. 3.
Fig. 3.
Protein–protein interaction network of the 89 biomarker proteins. Nodes represent proteins, while edges indicate predicted functional interactions based on STRING-db analysis. Node colors correspond to phenotype classifications derived from the Monarch Initiative, a database that integrates genotype–phenotype relationships: language impairment (red), neurological speech impairment (blue), and neurodevelopmental abnormality (yellow). This visualization highlights clusters of related biomarkers and shared molecular pathways relevant to developmental language disorders.
In addition to this qualitative visualization, we conducted a functional enrichment analysis of the network gene set against phenotype annotations from the Monarch Initiative. The major phenotype classes represented among the biomarker proteins directly relevant to developmental language disorders (DLDs) include Language impairment (in red), Neurological speech impairment (in blue), and Neurodevelopmental abnormality (in yellow). All of these enriched phenotype associations remained significant after correction for multiple testing (with FDR values on the order of 10-26 to 10-28), underscoring the robustness of the enrichment. The full spectrum of significant phenotype terms is summarized in Fig. 4.
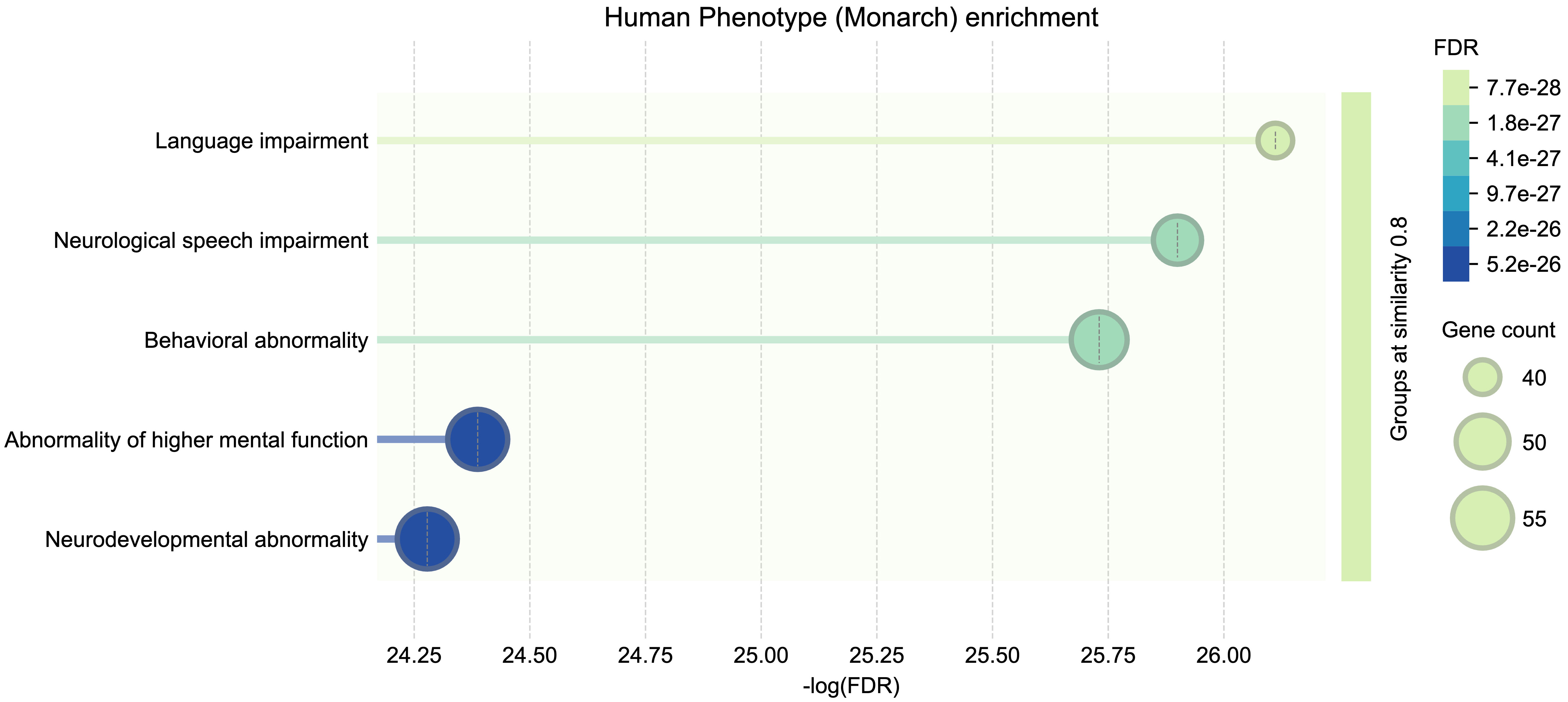 Fig. 4.
Fig. 4.
Functional enrichment analysis of the protein–protein interaction network. Phenotypic classifications are based on annotations from the Monarch Initiative, which integrates genotype–phenotype relationships. This visualization illustrates the enrichment of language-related and neurodevelopmental phenotypes among the identified biomarker proteins.
This analysis confirmed that the 89 genes are highly enriched for multiple phenotypic categories related to speech and language development. The STRING-based PPI analysis not only reinforced known connections among the genes but also uncovered previously unrecognized interactions. At the same time, the network revealed indirect linkages between biomarkers that were not evident from the Open Targets data alone, suggesting additional shared molecular pathways underlying DLDs. Together, these network results provide a more comprehensive view of the molecular network architecture underlying developmental language disorders.
The analysis of sequence and structural similarities among genes implicated in DLDs revealed only a few notable homology relationships, emphasizing their genetic heterogeneity. Among the genes surveyed, FOXP2 stands out as sharing significant sequence identity and conserved domain architecture with its paralog FOXP1. Both genes encode forkhead-domain transcription factors, and their DNA-binding (forkhead) domains are highly conserved (with over 90% amino acid identity). This high sequence similarity translates into a closely overlapping three-dimensional structure for the FOXP and FOXP1 forkhead domains, as expected given their common evolutionary origin. Similarly, CNTNAP2 (contactin-associated protein-like 2) exhibits strong sequence and structural homology with CNTNAP5, as both are members of the neurexin superfamily. These two large membrane proteins share a characteristic arrangement of extracellular domains—including multiple laminin G repeats and epidermal growth factor (EGF)-like domains—reflecting a preserved domain organization that likely underpins similar roles in neural cell adhesion and communication.
In contrast to these examples of paralogous gene pairs, the majority of DLD-associated genes show little to no sequence or structural similarity to one another. Even genes that converge on similar neurodevelopmental processes, such as ZNF277 (a zinc-finger protein implicated via a chromosomal inversion) and AUTS2 (a gene linked to neurodevelopmental regulation), do not exhibit recognizable sequence homology or protein structural alignment with each other or with the aforementioned gene families. This lack of commonality underscores the diverse molecular landscape of DLD risk genes, suggesting that multiple distinct biological systems are involved in language development and can be disrupted to produce the DLD phenotypes.
To explore potential hidden relationships, we performed a broad computational comparison of protein structures for all DLD-associated genes. We conducted pairwise sequence similarity analysis among the 89 proteins using amino acid sequence alignment with BLAST [83]. To assess the significance of the sequence identity scores, we followed Rost’s classification [84], which considers sequence identities above 30% as significant, indicating a high likelihood of homology. We compared these findings with structural similarity derived from 3D structural alignment, measured by the template modeling score (TM-score) [85]. A TM-score above 0.5 suggests that two proteins likely adopt the same fold and share an evolutionary relationship [86], while a score above 0.7 indicates strong structural similarity. While sequence similarity often predicts structural similarity, distinct sequences can still converge on similar structures and perform analogous functions, as protein folds are more evolutionarily conserved than sequences [87].
WDR45 and GNB1 exemplify this phenomenon: they exhibited significant structural similarity using their AlphaFold models (TM-score above 0.7), despite sharing only 5% sequence identity. Their structural alignment is shown in Fig. 5. This underscores the importance of looking beyond primary sequence similarity when studying gene function and evolution. Structural comparisons can reveal hidden relationships between proteins that may not be evident from sequence data alone. This is particularly relevant for DLDs, where functionally convergent genes may influence similar neurodevelopmental pathways despite lacking obvious sequence homology. By identifying shared folds or domain architectures, we can better predict protein interactions, infer potential molecular mechanisms, and prioritize genes for functional follow-up studies. Ultimately, integrating sequence and structure helps build a more complete picture of how diverse genetic risk factors contribute to the biology of language development and underscores that functionally related DLD genes might be discovered through structural homology even when sequence similarity is absent.
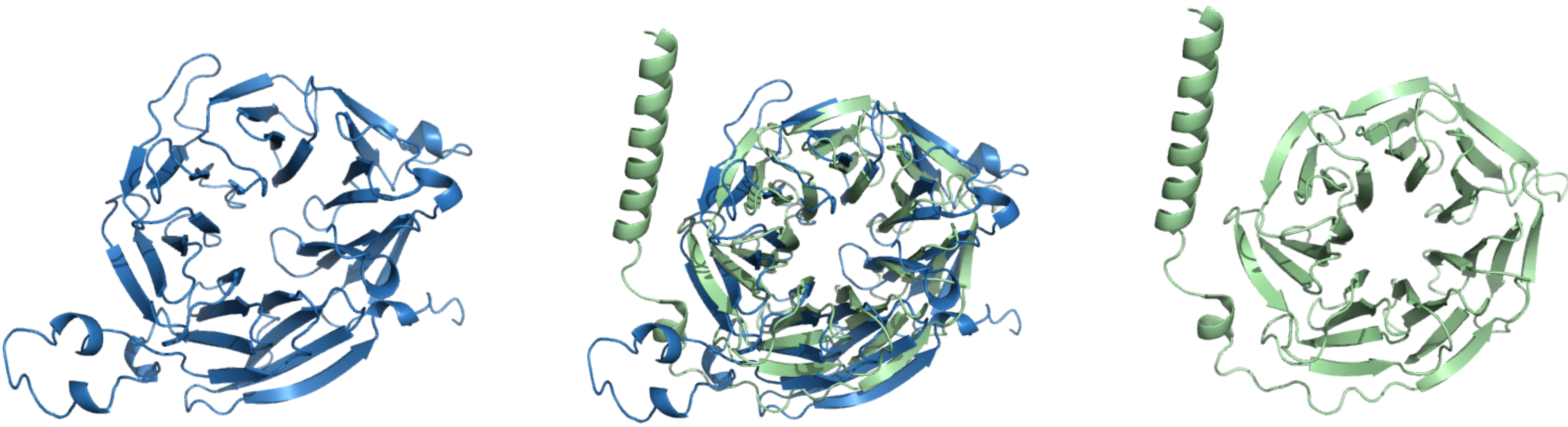 Fig. 5.
Fig. 5.
3D structures of WDR45 (blue, left) and GNB1 (green, right), with their structural alignment shown in the center.
The introduction of AlphaFold 3 [32] has significantly advanced structural biology by providing high-accuracy predictions of protein complexes, creating new opportunities in genomic research. Recognized with the 2024 Nobel Prize in Chemistry, AlphaFold 3 enables multimeric complex modeling with high accuracy, particularly benefiting biomarker interaction studies in different disorders. Such predictions provide a structural hypothesis that requires experimental validation to confirm their functional relevance.
One example is the potential structural interaction between KIAA0319 and DCDC2, two genes implicated in reading and language disorders. Previous studies suggested a genetic interaction between these genes in dyslexia [88, 89], though the precise molecular mechanism remains unclear. Using AlphaFold 3, we predicted the multimeric complex of KIAA0319 and DCDC2 (Fig. 6) to visualize potential interacting residues. While the strength and functional relevance of this interaction require experimental validation, structural predictions allow speculation on possible binding interfaces. Tentatively, if certain residues involved in this interaction coincide with known risk alleles, this could provide insights into how genetic variants may disrupt protein interactions and contribute to language-related disorders.
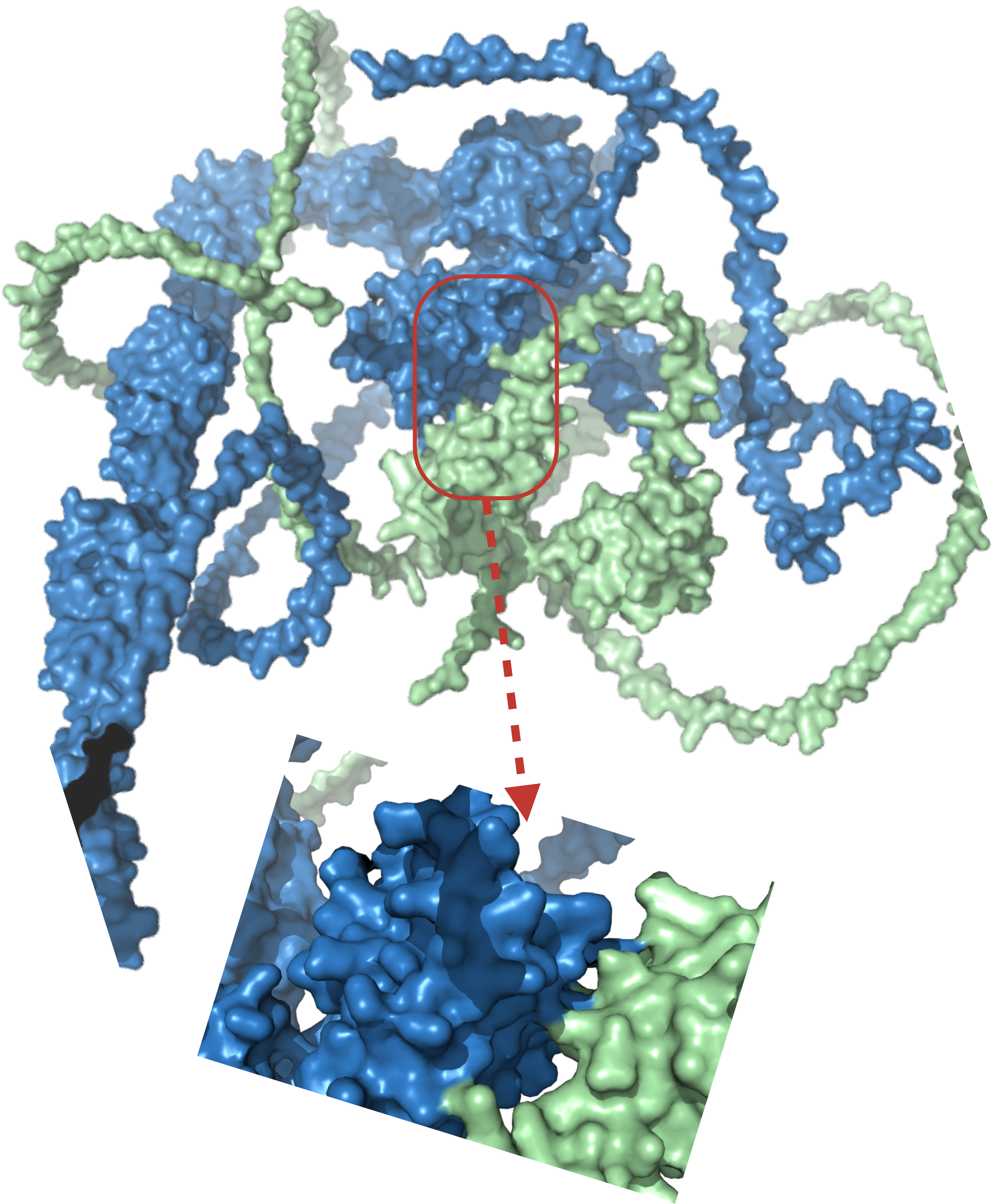 Fig. 6.
Fig. 6.
Predicted structural interaction between DCDC2 (green, UniProt ID: Q9UHG0) and KIAA0319 (blue, UniProt ID: Q5VV43) using AlphaFold 3 (v3.0.1; https://github.com/google-deepmind/alphafold3).
Pragmatically, while these structural predictions offer a visual representation of a potential interaction, they remain speculative and require experimental validation to confirm any functional significance. AlphaFold predicts a plausible binding interface between KIAA0319 and DCDC2, identifying specific residues at the interaction surface. Investigating known variants at these sites may reveal mechanisms by which genetic alterations disrupt this interaction, potentially contributing to the pathophysiology of language-related disorders.
A good biomarker is often a good therapeutic target. To our knowledge, there are currently no FDA- or EMA-approved drugs specifically for treating language disorders. However, several language-related biomarkers identified in our analysis are already targeted by approved drugs for neurological and psychiatric conditions. This suggests their potential utility in indirectly enhancing language function or modulating underlying cognitive mechanisms.
Among the 89 genes analyzed, several have FDA-approved drugs, emphasizing their translational relevance (see Supplementary Table 1):
• GRIN Family (Glutamate Receptor, Ionotropic, N-Methyl D-Aspartate)—These NMDA receptor subunits (GRIN1, GRIN2A-D, GRIN3A-B) are targeted by drugs approved for depression, autism, and amyotrophic lateral sclerosis. Their role in synaptic plasticity makes them compelling targets for modulating cognitive functions related to language.
• ACHE—Targeted in approved treatments for Alzheimer’s disease, Parkinson’s disease, and memory impairment. ACHE inhibitors have been FDA-approved since the 1950s and are foundational in neurodegenerative disease management.
• DRD2 & DRD3 (Dopamine Receptors)—These are among the most extensively targeted psychiatric biomarkers, with drugs approved for schizophrenia, bipolar disorder, depression, and psychosis. DRD2, in particular, is one of the most validated central nervous system (CNS) drug targets.
• HTR2A (Serotonin Receptor 2A)—Implicated in mood regulation and cognitive flexibility, with approved drugs for schizophrenia, anxiety, obsessive-compulsive disorder, and depression. These functions intersect with cortical language processing.
• GABBR1 (GABA-B Receptor Subunit 1)—Targeted by drugs for hypersomnia and spinal cord diseases. Although not directly associated with language disorders, GABAergic modulation may influence neural inhibition mechanisms that are important in speech and language processing.
• HDAC8 (Histone Deacetylase 8)—Targeted in cancers such as multiple myeloma and Burkitt lymphoma. Its involvement in epigenetic regulation suggests potential relevance to neural plasticity and development.
• SCN Family (voltage-gated sodium channel genes)—These sodium channel genes (e.g., SCN1A, SCN2A, SCN9A) are implicated in epilepsy and pain. Given the frequent co-occurrence of epilepsy and language impairment, these targets may offer indirect therapeutic relevance.
• NR3C1 (Glucocorticoid Receptor)—Targeted in a range of inflammatory and autoimmune disorders. Chronic stress and dysregulation of glucocorticoid signaling can impair cognitive and language development, positioning this as a target of interest.
The existence of FDA-approved drugs for these targets highlights opportunities for drug repurposing and accelerates the translational pathway from biomarker identification to therapeutic application. These genes may serve not only as diagnostic or prognostic biomarkers but also as modulators of therapeutic response in future precision medicine approaches for language disorders.
Unlike diseases such as cancer, where specific genomic panels like Oncotype DX and MammaPrint guide clinical decision-making, genetic testing for developmental language disorders (DLDs) is not yet a standard practice. However, in cases where DLDs coexist with other neurodevelopmental anomalies or where a family history of genetic syndromes suggests an underlying genetic cause, genetic testing may be warranted. Several commercially available genetic tests are used in the clinical assessment of neurodevelopmental disorders, including DLDs, autism spectrum disorder (ASD), and intellectual disability (ID). These tests help identify genetic variants associated with language impairments, contributing to better diagnosis, personalized interventions, and risk assessment for affected individuals and their families.
(1) Chromosomal microarray analysis (CMA) [90] is a genetic test commonly used for individuals with developmental delays, intellectual disabilities, or congenital anomalies. It detects copy number variations (CNVs), microdeletions, and duplications that may contribute to neurodevelopmental disorders. However, CMA does not detect single-gene mutations or small sequence variations. Several commercially available CMA-based tests are used in clinical practice. One example is Affymetrix CytoScan® Dx Assay, the first CMA test to receive FDA approval for the genetic evaluation of individuals with developmental delay [91].
(2) Next-generation sequencing (NGS) panels for neurodevelopmental disorders provide a comprehensive approach by sequencing multiple genes simultaneously, identifying genetic variants associated with neurodevelopmental conditions. Several targeted gene panels include genes implicated in speech and language disorders, such as FOXP2 and KIAA0319. Commercially available NGS panels include:
• Invitae Neurodevelopmental Disorders Panel [92, 93], a targeted sequencing panel covering hundreds of genes associated with neurodevelopmental disorders.
• Ambry Genetics Neurodevelopmental Disorders Panel [94].
• Blueprint Genetics Autism Spectrum Disorders Panel [95].
Additionally, CENTOGENE offers specialized NGS panels [96] for diagnosing hereditary neurological disorders, covering conditions such as ataxia, epilepsy, mitochondrial diseases, and neuromuscular disorders. Key panels include:
• Ataxia/Spastic Paraplegia Panel (483 genes).
• CentoICU® (856 genes for critically ill infants).
• CentoMito® Comprehensive (451 genes for mitochondrial disorders).
• CentoNeuro (1902 genes covering broad neurological conditions).
• Epilepsy Panel (783 genes).
While these genetic tests provide valuable insights into the genetic architecture of DLDs, their clinical utility remains limited due to several challenges:
• Variant interpretation—many identified variants are classified as variants of uncertain significance (VUS), complicating clinical decision-making.
• Polygenic nature of DLDs—no single genetic test can fully explain its etiology.
• Neurodevelopmental overlap—language disorders share genetic risk factors with autism spectrum disorder (ASD), attention deficit hyperactivity disorder (ADHD), and intellectual disabilities (ID).
• Genetic syndromes with language deficits—some genetic syndromes, such as Williams syndrome, Fragile X syndrome, and FOXP1-related disorders, include language impairments as a core feature.
• Terminological inconsistencies: Research on language disorders faces challenges related to terminology, heterogeneity, and underlying complexity. A major issue is the lack of standardized diagnostic criteria. The CATALISE Consortium proposed “developmental language disorder (DLD)” as a unified term for idiopathic language impairment [17, 97], and the International Classification of Diseases, 11th Revision (ICD-11) for Mortality and Morbidity Statistics [55] categorizes DLD. However, inconsistent definitions persist, hindering cross-study comparisons and clinical applications, which further complicates our research.
• Symptom and phenotypic heterogeneity: Children with language disorders present a wide range of symptoms, which can vary across individuals, languages, and environments. Cross-linguistic differences mean that a language impairment might manifest differently depending on the linguistic context (for instance, grammar-rich languages vs. more analytical languages). Socio-environmental factors such as socioeconomic status and language exposure also influence the severity and outcomes of DLD. This high variability makes it challenging to define clear diagnostic boundaries and to identify consistent phenotypes for genetic studies.
• Complex gene–environment interplay and non-specific genetic markers: The genetic architecture of DLD is highly complex and polygenic. Many different genes of small effect interact with each other and with environmental influences, so disentangling innate predisposition from external modulation is difficult. Moreover, many genetic markers implicated in DLD are not unique to language disorders—they overlap with broader neurodevelopmental conditions like autism and ADHD. This complexity highlights the challenge of distinguishing a “primary” language disorder from language difficulties that are part of a broader syndrome. In short, few genetic findings are specific to DLD, and any given risk variant often contributes to multiple developmental outcomes rather than a single, isolated language deficit.
Developmental language disorders (DLDs) do not stem from a single gene or simple cause. Our review underscores the complex, polygenic nature of DLDs: many different genes contribute, each adding a modest risk. For example, we found that both specialized language-related genes (like FOXP2) and broader neurodevelopmental genes (such as DCDC2 or GRIN2B) are implicated. No one pathway drives DLD; instead, these diverse genetic factors interact and often affect multiple aspects of development beyond language. By analyzing gene networks and protein structures, we identified shared biological pathways that link many DLD-associated genes. Although advanced tools like large-scale DNA sequencing and AI-driven protein modeling have begun to illuminate how DLD genes function, much of the genetic puzzle remains unsolved—many findings are still unexplained, and we do not yet fully understand how most genetic variants lead to language difficulties. Nevertheless, the growing knowledge offers hope for earlier diagnosis and personalized intervention. If reliable genetic biomarkers can be identified, children at risk for DLD might be recognized earlier in life, allowing for timely and targeted support. A genetic profile could one day help tailor interventions to the needs of each child, improving the effectiveness of therapies.
The data presented in this study are contained in this article are available upon request from the corresponding author.
RAH and AA conceived the study and designed the methodology. RAH performed the data analysis, with input from AA. RAH contributed to data curation and validation. AA supervised the study. RAH wrote the manuscript. Both authors contributed to editorial changes in the manuscript. Both authors read and approved the final manuscript. Both authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
The authors thank the anonymous reviewers for their valuable suggestions.
This research received no external funding.
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBS38706.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.