
- Academic Editor
†These authors contributed equally.
Heterotaxy syndrome is characterized by abnormal organ arrangement across the left-right (L-R) axis, often leading to complex congenital heart defects (CHDs). Genetic analysis via whole-exome sequencing revealed two novel compound heterozygous mutations in the polycystic kidney disease 1 like 1 (PKD1L1) gene (NM_138295.3: c.6659T>A and c.8104dup). These genetic alterations are implicated in the abnormal development of the L-R axis, contributing to the severe cardiac malformations observed.
This case report describes a Chinese fetus diagnosed with heterotaxy and severe cardiac anomalies identified through prenatal ultrasound.
Our results expand the known spectrum of PKD1L1 mutations and highlight the importance of genetic testing in prenatal diagnosis of heterotaxy. These findings emphasize the value of genetic testing in informing clinical decisions and guiding reproductive counseling.
The human heart and internal organs are typically categorized into three configurations: situs solitus, situs inversus (including Kartagener syndrome), and heterotaxy [1, 2]. Situs solitus represents the standard anatomical arrangement, with all major organs in their expected positions. Situs inversus totalis (SIT) involves a complete mirror-image reversal of the viscera. Although overall health often remains unaffected with SIT due to the organ relational concordance being maintained, this reversal can pose significant challenges in medical diagnostics and interventions [3].
In contrast, heterotaxy syndrome presents significant clinical challenges due to its randomized and often discordant visceral arrangement. This condition arises from disruption in the normal differentiation of the left-right (L-R) axis during embryonic development, resulting in severe congenital heart malformations in approximately 80% of affected cases [4]. Differentiation of the L-R axis is intricately linked to ciliary motion and nodal flow. Cilia are hair-like structures on cell surfaces that generate a leftward fluid flow at the embryonic node. This nodal flow is crucial for establishing the correct L-R axis and guiding the symmetrical placement of organs [5]. A number of genes have been implicated in heterotaxy, including activin a receptor type iib (ACVR2B), cripto, frl-1, cryptic family 1 (CFC1), nodal growth differentiation factor (NODAL), cilia and flagella associated protein 52 (CFAP52), zic family member 3 (ZIC3), nephronophthisis 2 (NPHP2), nephronophthisis 3 (NPHP3), nephronophthisis 2 (NPHP4), polycystic kidney disease 2 (PKD2), tetratricopeptide repeat domain 8 (TTC8), and PKD1L1. The exact mechanisms by which these genes influence differentiation of the L-R axis remains only partially understood, with some genes encoding proteins related to ciliary function rather than structural components [6, 7].
The PKD1L1 gene, located on chromosome 7p12.3, is crucial for L-R axis patterning [8, 9]. This is evidenced by studies in homozygous mutant mice (Pkd1l1–/-), which commonly display situs inversus without exhibiting symptoms typically associated with primary ciliary dyskinesia, indicating that PKD1L1 likely acts downstream of nodal flow [9]. Additional evidence for the role of PKD1L1 in this process comes from studies in medaka fish, where mutations in PKD1L1 lead to randomized cardiac looping and dislocation of the liver and gallbladder [10]. Corresponding bi-allelic mutations in humans have similarly been linked to heterotaxy syndrome, further underscoring the significant role of PKD1L1 genes in developmental patterning [7].
This study presents the case of a fetus diagnosed with heterotaxy of the heart
and carrying novel compound heterozygous mutations in PKD1L1
(c.6659T
This study was approved by the Clinical Research Ethics Committee of The Second Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, China. Written informed consent was obtained from the parents for the collection of their blood and of aborted fetal samples. The parents agreed to publish the clinical and genetic data relevant to this case. Genomic DNA was extracted using the MagPure Buffy Coat DNA Midi KF Kit (Magen Bio, Guangzhou, Guangdong, China), following the protocol provided by the manufacturer.
Genomic DNA was fragmented (150–250 bp) using Covaris High-throughput Sequencing Set (Covaris, Inc., Woburn, MA, USA) and purified. It then underwent end-repair, 3’ adenylation, adapter ligation, and polymerase chain reaction (PCR). PCR products were purified and hybridized using BGI Hybridization and Wash Kits (BGI, Shenzhen, Guangdong, China). Following purification, the PCR products were denatured, circularized into single-strand circular DNA, and quality-controlled. This library was amplified to form DNA nanoballs (DNBs). Sequencing was performed on the MGISEQ-2000 High-throughput Sequencing Set (MGI, Shenzhen, Guangdong, China) platform using the combinatorial Probe-Anchor Synthesis (cPAS) method, which covers the exonic regions of approximately 20,000 human genes.
Raw data were processed to remove low-quality and adapter-contaminated reads. High-quality reads were aligned to the human reference genome (GRCh37/hg19) using the Burrows-Wheeler Aligner (BWA, v0.7.17, Wellcome Sanger Institute, Hinxton, Cambridgeshire, UK) [11]. Genome Analysis Toolkit (GATK v4.4.0.0, Broad Institute, Cambridge, MA, USA) [12] was used for recalibration, variant calling, and filtering. Variants were annotated using SnpEff (v.5.1d, Pablo Cingolani, La Jolla, CA, USA) [13] based on RefSeq annotations. Allele frequencies were sourced from the 1000 Genomes Project (https://www.internationalgenome.org/), The Genome Aggregation Database (gnomAD) (https://gnomad.broadinstitute.org/), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), and Database for Nonsynonymous SNPs’ Functional Predictions (dbNSFP) (https://sites.google.com/site/jpopgen/dbNSFP) databases. The potential functional impacts of amino acid substitutions were assessed using Tolerant (SIFT) [14] and PolyPhen2 [15]. PCR and Sanger sequencing were utilized to confirm variants in the parents and aborted fetus.
Genomic DNA was enzymatically digested and end-repaired with an adenine (A) base. Adapters containing tag sequences were then ligated to both ends of the DNA fragments. The library was prepared by PCR amplification and subsequent purification, followed by single-stranded circular (ssCir) DNA and rolling circle replication to form DNBs for sequencing.
Sequencing data were processed by aligning to a reference genome (GRCh37/hg19), with subsequent deduplication, GC content, and window adjustment. Analysis of CNV was performed using the Population-Scale CNV Calling (PSCC) [16] method, incorporating rigorous quality control measures. Variant annotation was performed using PKD1L1 transcript NM_138295.3 (RefSeq accession from National Center for Biotechnology Information (NCBI) Gene). Identified variants were annotated and classified semi-automatically utilizing databases such as Online Mendelian Inheritance in Man (OMIM) (https://www.omim.org/), Human Gene Mutation Database (HGMD) (https://www.hgmd.cf.ac.uk/ac/index.php), and Database of Genomic Variants (DGV) (https://dgv.tcag.ca/dgv/app/home) to facilitate accurate interpretation and reporting. The resolution of CNV-seq analysis exceeds 100 kilobases (kb).
A 28-year-old pregnant Chinese woman presented at our clinic for evaluation after obstetric ultrasound at 18 weeks of gestation had revealed complex congenital heart defects in the fetus. This was her first pregnancy, and she stated that she had not been exposed to radioactive substances or toxins. Additionally, there were no elevated risk indicators for fetal nuchal translucency or signs of Down syndrome during early and second-trimester screenings. Both parents exhibited normal phenotypes and there was no known family history of congenital heart defects or laterality disorders. A follow-up ultrasound at our hospital confirmed multiple fetal anomalies, including heart malformations, duodenal obstruction, endocardial cushion defect (ECD), ventricular septal defect (VSD), and single atrium (SA) (Fig. 1). No abnormalities were detected in the fetal liver, spleen, or stomach. The parents opted for termination of the pregnancy and sought reproductive counseling. They also gave consent to include their data in this research report.
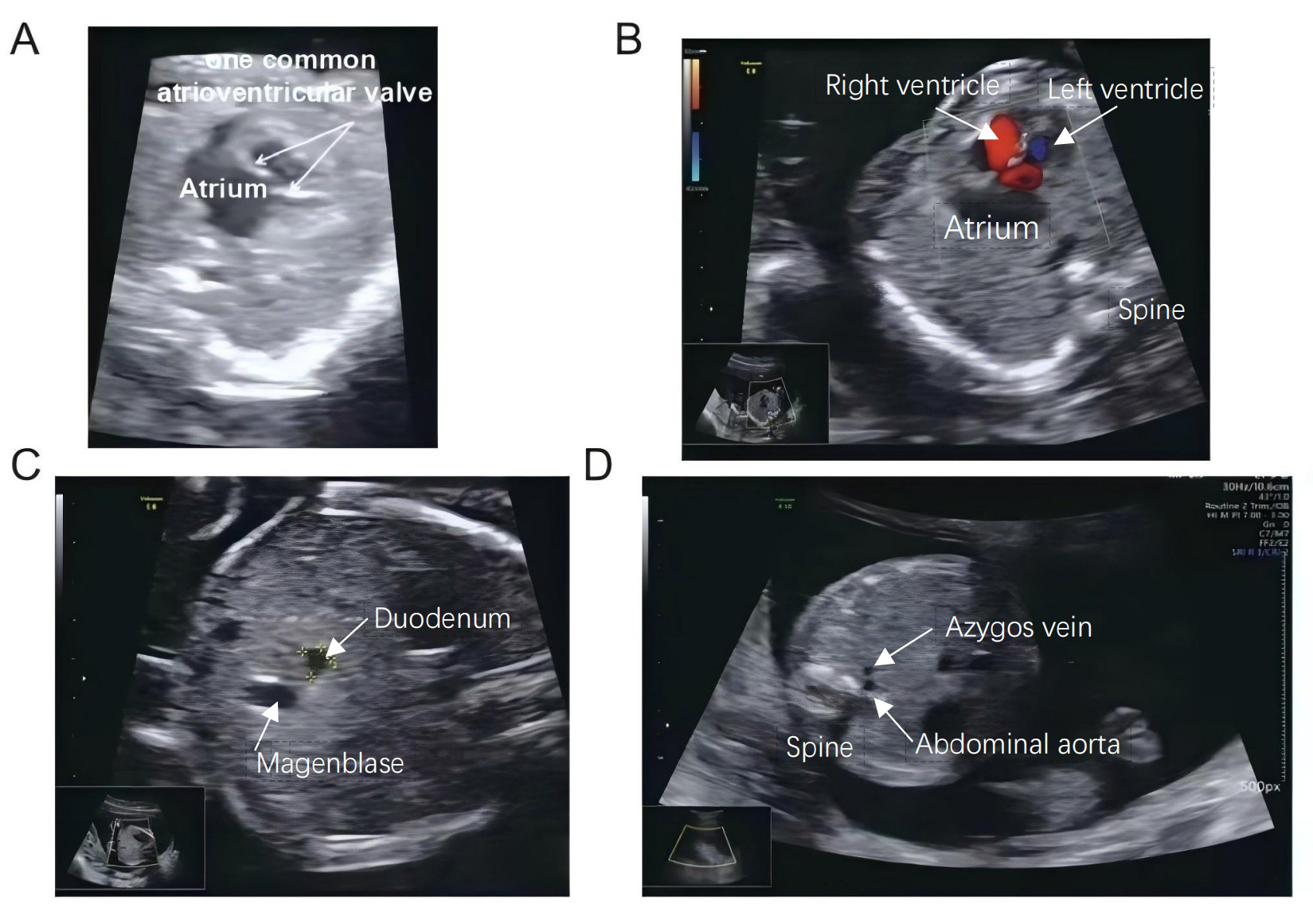 Fig. 1.
Fig. 1.
Ultrasound and echocardiographic findings in fetus with congenital heart disease. (A) Ultrasound examination revealed a single common atrioventricular valve in the fetal heart. (B) Echocardiography shows ventricular septum interruption measuring approximately 0.18 cm. (C) Image illustrates a dilated duodenum adjacent to the abdominal aorta in the peritoneal plane. (D) A dilated azygos vein is also evident next to the abdominal aorta. The upper white arrow in panel C indicates the dilated duodenum, while the yellow markers show the margin of the duodenum.
CNV-seq and WES were conducted on DNA extracted from the aborted fetus. The CNV-seq analysis showed no chromosomal aneuploidies or CNVs. However, WES analysis revealed novel compound heterozygous mutations in the PKD1L1gene in the fetus (Fig. 2). Reports of pathogenic mutations in PKD1L1 are rare, with only a limited number of both homozygous and heterozygous pathogenic mutations documented in various populations.
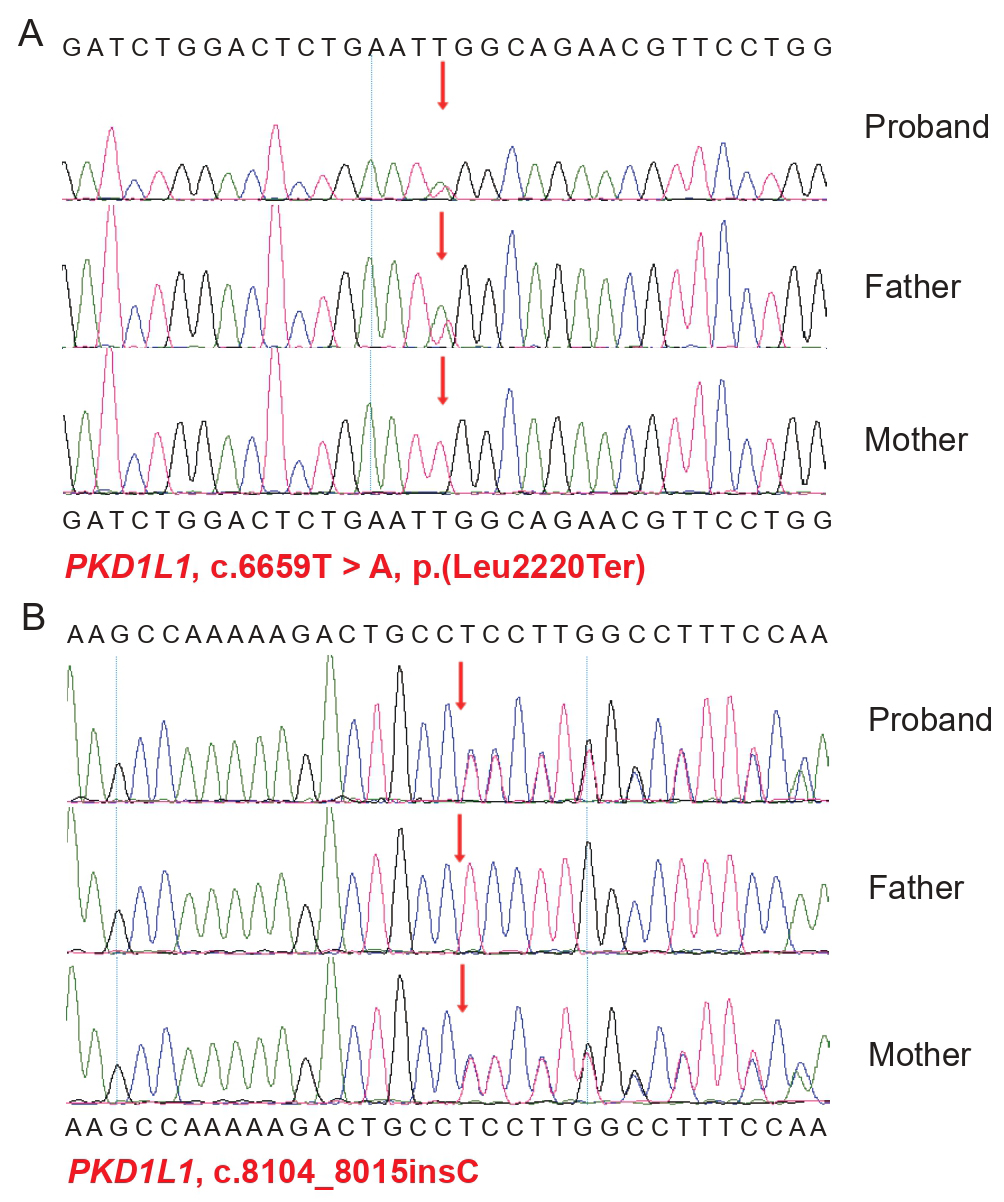 Fig. 2.
Fig. 2.
Sanger sequencing results showing PKD1L1 mutations in
the fetus and its parents. Arrows indicate the location of the mutations. Sanger
sequencing confirmed the fetus carried heterozygous mutations in the PKD1L1 gene. (A) c.6659T
The compound heterozygous mutations identified in the PKD1L1 gene are
located in exons 44 and 54, respectively. The c.6659T
The PKD1L1 protein comprises six essential structural domains: Polycystic kidney disease domains (PKD) domains, Receptor for Egg Jelly (REJ), G Protein-coupled Receptor Proteolytic Site (GPS), Polycystin-1, Lipoxygenase, Alpha-Toxin/Lipoxygenase Homology 2 (PLAT/LH2), and coiled-coil domains, as well as the transmembrane regions. Together, these domains contribute to the functionality of PKD1L1 in cellular adhesion, calcium transport, protein interactions, and intracellular signaling [8].
To further explore the relationship between PKD1L1 gene mutations and heterotaxy, we compiled the reported pathogenic PKD1L1 mutations associated with heterotaxy phenotypes. Table 1 (Ref. [6, 7, 17, 18, 19, 20, 21, 22, 23, 24]) provides a summary of published clinical reports on PKD1L1-related heterotaxy, detailing the clinical features, genetic spectrum (including information on variant positions), and patient populations.
| No. | Clinical Features | CDS change | Protein change | PKD1L1 Domain | Population | Ref |
| 1 | Situs ambiguous, AVSD, left ventricular hypoplasia, malposed arteries | c.6473+2_6473+3delTG | Splicing variant | / | Northern European | [7] |
| 2 | SIT, pulmonary atresia, VSD | c.5072G |
p.Cys1691Ser | GPS | Iranian | [7] |
| 3 | Heterotaxy, left bronchial isomerism, malposed arteries | c.850C |
p.Arg284* | / | European | [17] |
| c.7579C |
p.Gln2527* | / | ||||
| 4 | Situs solitus, L atrial isomerism, bilateral SVC, VSD, CoA, malrotated intestines | c.4039C |
p.Arg1347* | REJ | European | [18] |
| c.4798_4799del | p.Gln1600Aspfs*4 | / | ||||
| 5 | Situs anomaly | c.5072G |
p.Cys1691Ser | GPS | Unknown | [19] |
| 6 | SIT, anemia | c.8005C |
p.Arg2669* | / | South Asian | [20] |
| 7 | Visceral situs inversus, dextrocardia, intestinal malrotation, hydrops | c.8005C |
p.Arg2669* | / | Indian | [21] |
| c.160+1G |
Splicing variant | / | ||||
| 8 | Bronchial asthma, membranous duodenal stenosis, gastroesophageal reflux | c.5660C |
p.Thr1887Met | PLAT/LH2 | European | [22] |
| c.4019_4033dup | p.Gln1344_Trp1345insSerSer CysAsnGln | REJ | ||||
| 9 | Heterotaxy, congenital asplenia | c.1387C |
p.Gln463* | / | Chinese | [6] |
| 10 | Chylothorax, hydrops fetalis, persistent pulmonary hypertension, respiratory failure | c.1543G |
p.Gly515Arg | PKD 1 | Unknown | [23] |
| c.3845T |
p.Val1282Glu | REJ | ||||
| 11 | Hydrothorax, hydrops fetalis, severe pulmonary hypoplasia, persistent pulmonary hypertension, cardio-respiratory failure | c.863delA | p.Asn288Thrfs*3 | / | Unknown | |
| c.6549G |
p.Gln2183His | / | ||||
| 12 | SIT | c.7663C |
p.Arg2555* | Coiled-coil | Turkish | [24] |
| c.7937C |
p.Ser2646* | Coiled-coil | ||||
| 13 | Heterotaxy, SA, VSD, ECD | c.6659T |
p.Leu2220* | / | Chinese | Present study |
| c.8104dup | p.Leu2702Profs*8 | / |
*, termination codon; AVSD, atrioventricular septal defect; SIT, situs inversus totalis; VSD, ventricular septal defect; SVC, superior vena cava; CoA, coarctation of the aorta; SA, single atrium; ECD, endocardial cushion defect; PKD 1, Polycystic kidney disease domains 1; REJ, Receptor for Egg Jelly; GPS, G Protein-coupled Receptor Proteolytic Site; PLAT/LH2, Polycystin-1, Lipoxygenase, Alpha-Toxin/Lipoxygenase Homology 2; Ref, reference.
The reported pathogenic PKD1L1 mutations associated with heterotaxy include five missense mutations (p.Cys1691Ser, p.Thr1887Met, p.Gly515Arg, p.Val1282Glu, p.Gln2183His) and an in-frame insertion (p.Gln1344_Trp1345insSerSerCysAsnGln). The p.Gln2183His mutation is located in the transmembrane region, which is crucial for anchoring the protein and supporting its functional activities at the cellular membrane.
The other five variants are located within critical functional domains of the PKD1L1 protein: GPS, PLAT/LH2, PKD 1, and REJ. Mutations in these regions can disrupt protein conformation and stability, potentially compromising normal functions. Disruptive mutations, which include nonsense, frameshift and splicing mutations, were more prevalent than milder missense mutations (Table 1) (Ref. [6, 7, 17, 18, 19, 20, 21, 22, 23, 24]). Such variants are severe and are likely to result in a significant loss of protein function.
A review of the clinical features in patients with PKD1L1 mutations reveals a spectrum of symptoms, emphasizing the critical role of this gene in L-R orientation. Conditions such as pulmonary atresia, atrial isomerism, and dextrocardia highlight the influence of PKD1L1 on cardiac and pulmonary positioning, linking it directly to heterotaxy syndrome. Furthermore, its association with intestinal malrotation and other visceral malformations extends its impact to overall organ orientation.
In the present case, both mutations found in the PKD1L1 gene,
c.6659T
This study has the following limitations: (1) Due to constraints in clinical samples, functional validation of the PKD1L1 variants through cellular experiments was not performed; (2) The absence of an autopsy (declined by the family) precluded pathological confirmation of visceral situs abnormalities. These factors may affect the accuracy of genotype-phenotype correlations.
In summary, our study found two novel PKD1L1 gene mutations
(c.6659T
Considering both parents are carriers of a pathogenic variant, the recurrence risk for future pregnancies is 25%. It’s crucial to counsel the parents about this risk and the availability of prenatal diagnosis and preimplantation genetic testing to guide future reproductive decisions.
The data that support the findings of this study have been deposited into CNGB Sequence Archive (CNSA) of China National GeneBank DataBase (CNGBdb) (https://db.cngb.org/cnsa/) with accession number CNP0005720.
XT and JZ designed this study and wrote the first draft of the paper. JY collected the ultrasound data. JS, LT, JX and YC conducted the literature search and review, revised the manuscript. LW made substantial contributions to the design of the study, supervised the research and reviewed the final manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
All subjects gave their informed consent for inclusion before they participated in the study. The study was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the Ethics Committee of The Second Affiliated Hospital, School of Medicine, Zhejiang University (approval number: 2024-0644).
We would like to express our gratitude to all those who helped us during the writing of this manuscript. Thanks to all the peer reviewers for their opinions and suggestions.
This project was supported by the Zhejiang Provincial Natural Science Foundation (Grant No.: LMS25H040003).
The authors declare no conflict of interest. Liquan Wang is serving as one of the Editorial Board members of this journal. We declare that Liquan Wang had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Michael H. Dahan.
During the preparation of this work the authors used Deepseek in order to check spelling and grammar. After using this tool, the authors reviewed and edited the content as needed and takes full responsibility for the content of the publication.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/CEOG41691.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.