







 , Thu Minh Phung 1,3,†, Anh Thi Mai Do 1, Quynh Tran Mai Ly 4, Tin Hoang Nguyen 1,2,*
, Thu Minh Phung 1,3,†, Anh Thi Mai Do 1, Quynh Tran Mai Ly 4, Tin Hoang Nguyen 1,2,*
1 Faculty of Medicine, Can Tho University of Medicine and Pharmacy, 900000 Can Tho, Vietnam
2 Department of Functional Exploration, Can Tho University of Medicine and Pharmacy Hospital, 900000 Can Tho, Vietnam
3 Department of Pathology, Can Tho University of Medicine and Pharmacy Hospital, 900000 Can Tho, Vietnam
4 Faculty of Pharmacy, Can Tho University of Medicine and Pharmacy, 900000 Can Tho, Vietnam
†These authors contributed equally.
Abstract
This review was conducted to explain how menopausal hormone therapy (MHT) benefits cardiovascular diseases (CVDs) and how to control the risk of breast cancer.
Estrogen deficiency, altered energy homeostasis, adipocyte changes, inflammation, and insulin resistance are responsible for the development of metabolic syndrome and CVDs. Estrogen influences hypothalamic function and maintains the energy balance, protecting menopausal women from these cardiovascular risk factors. However, estrogen metabolism plays a crucial role in the genotoxic pathway that leads to breast cancer. Moreover, MHT is associated with cell proliferation and mutation signaling pathways in breast cancer, as well as the process of growing the breast cancer stem cell.
While MHT may have favorable effects when started early, introducing it later in the course of atherosclerosis may pose major dangers, underlining the importance of timing in hormone therapy. Estrogen-only therapy has a greater favorable effect on CVDs than the estrogen-progesterone combination. Although the connection between MHT and breast cancer is well-documented, significant knowledge gaps remain, especially regarding the long-term effects of newer MHT formulations. Current studies support using the lowest effective dose for the shortest possible duration, with a focus on tailoring therapy to individual risk factors, such as obesity, smoking, and alcohol consumption. Thus, MHT should be customized due to the intricacy of individual risk factors and differences in responses to therapy.
Although MHT is effective for controlling CVDs in women entering menopause, it must be used with caution, especially in women at high risk of breast cancer.
Keywords
- atherosclerosis
- cardiovascular risk factors
- menopausal women
- obesity
- estradiol
- cell proliferation
- mutation
As individuals approach the average retirement age, typically between 50 and 70 years, they become more susceptible to a range of diseases, both mild and severe. In women, this period coincides with menopause—a natural biological transition marked by the cessation of ovarian hormone production, particularly estrogen and progesterone. The decline in these hormones is a significant contributor to the increased incidence of cardiovascular diseases (CVDs) among women over 55 years. Common cardiovascular conditions in this demographic include coronary heart disease (CHD), myocardial infarction (MI), heart failure, venous thromboembolism (VTE), and deep vein thrombosis [1, 2, 3, 4, 5].
Menopausal hormone therapy (MHT) has been proposed as a potential intervention to address the rising prevalence of CVDs among menopausal women. It is primarily used to relieve menopausal symptoms, such as hot flashes, night sweats, and vaginal dryness. It involves the administration of estrogen, and sometimes progesterone or progestin, to restore hormonal balance. While MHT can improve quality of life and prevent bone loss, it is not without risks, including an increased risk of blood clots, breast cancer, and heart disease—particularly for women who begin therapy after age 60 years or use it long-term. These risks have led to the cautious prescription of MHT, tailoring it to individual health needs and recommending the shortest effective duration [1, 6, 7]. Historically, MHT was widely used; however, after the Women’s Health Initiative (WHI) found an increased risk of CVDs, breast cancer, and stroke associated with MHT in 2004, its use declined significantly. More recent research, including several meta-analyses, has highlighted the potential protective effects of estrogen against CVDs, although the evidence remains controversial [8, 9, 10].
MHT impacts cardiovascular health by affecting lipid profiles, blood pressure, and glucose metabolism. Reduced estrogen levels during menopause increase low-density lipoprotein (LDL) cholesterol and triglycerides, reduce high-density lipoprotein (HDL) cholesterol, and elevate blood pressure, collectively raising the risk of atherosclerosis, MI, and stroke. Estrogen deficiency also impairs glucose tolerance and insulin sensitivity, increasing the risk of type 2 diabetes and cardiovascular complications. Research suggests that starting MHT within 10 years of menopause onset may mitigate these risks, but its safety in women over 60 years remains uncertain [5, 8, 9, 10].
In parallel, breast cancer risk has become a prominent concern associated with MHT. Epidemiological studies consistently demonstrate that the prolonged use of combined estrogen-progesterone therapy increases the risk of breast cancer, especially when therapy exceeds 3 to 5 years. Estrogen’s role in breast tissue involves stimulating cell proliferation and activating genotoxic pathways, which may contribute to malignancy. These findings emphasize the need to carefully balance the benefits of MHT for symptom relief and cardiovascular protection against its potential to elevate breast cancer risk [1, 11, 12].
While CVDs and breast cancer have distinct etiologies, estrogen deficiency emerges as a shared factor that links these conditions. Estrogen’s protective effects on cardiovascular health are diminished post-menopause, while altered estrogen metabolism contributes to carcinogenesis in breast tissue. This dual role underscores the complexity of prescribing MHT, which must consider individual risk profiles to optimize its benefits while minimizing harm.
This review aims to explore the impact of MHT on CVDs in menopausal women, focusing on its effects on lipid levels, blood pressure, glucose tolerance, and metabolic syndrome, particularly obesity. It also examines the mechanisms through which MHT may influence breast cancer risk. By addressing these areas, the review seeks to provide a nuanced understanding of how MHT can be used effectively and safely to manage menopausal health.
Menopause marks a critical transition in a woman’s life. It is characterized by the cessation of menstrual cycles and a significant decline in estrogen levels. This hormonal shift has profound effects on cardiovascular health, leading to both primary and secondary outcomes that heighten cardiovascular risk [5, 13, 14, 15]. Estrogen is integral in maintaining cardiovascular health through its influence on lipid metabolism, blood pressure regulation, and glucose tolerance. As estrogen levels decrease during menopause, these protective mechanisms weaken, resulting in notable adverse changes in cardiovascular risk factors [9, 10, 16, 17, 18]. One primary outcome of menopause is the alteration in lipid concentrations. Estrogen benefits lipid profiles by increasing HDL cholesterol, often referred to as good cholesterol, and decreasing LDL cholesterol, known as bad cholesterol. With reduced estrogen levels, women experience an unfavorable shift in lipid concentrations, characterized by an increase in LDL cholesterol and a decrease in HDL cholesterol [8, 15, 19]. This dyslipidemia accelerates the development of atherosclerosis, a condition in which plaque builds up in the arterial walls, leading to a higher risk of coronary artery disease and other cardiovascular events. Another frequent primary outcome of menopause is an increase in blood pressure [5, 14, 19, 20]. Estrogen contributes to blood pressure regulation through its effects on vascular function and sodium balance. As estrogen levels decline, systolic and diastolic blood pressure tend to rise. Elevated blood pressure, or hypertension, further contributes to the risk of CVDs such as heart attack, stroke, and heart failure [2, 10, 13, 21, 22]. The combination of increased blood pressure and adverse changes in lipid profiles creates a compounded risk for women during and after menopause. This heightened risk is partly due to the declining levels of estrogen, a hormone that plays a vital role in maintaining glucose metabolism. Estrogen enhances insulin sensitivity and supports glucose uptake in peripheral tissues, thereby helping to regulate blood sugar levels. During menopause, reduced estrogen levels can lead to insulin resistance, which increases the risk of impaired glucose metabolism and type 2 diabetes. Women undergoing menopause are more likely to experience severe cardiovascular conditions due to the cumulative effects of dyslipidemia, hypertension, and impaired glucose metabolism [19, 23, 24, 25]. The incidence of heart disease, which is lower in premenopausal women than in men, increases after menopause and can surpass that in men. To address these heightened risks, effective management strategies are crucial. Regular monitoring of lipid levels, blood pressure, and glucose tolerance is essential for early detection and intervention. Lifestyle modifications, such as adopting a heart-healthy diet, engaging in regular physical activity, and maintaining a healthy weight, are beneficial. Additionally, pharmacological treatments may be required to manage dyslipidemia, hypertension, and glucose intolerance [7, 23, 24, 26]. MHT has been considered a means to alleviate some menopausal symptoms and potentially improve cardiovascular outcomes, but its benefits must be weighed against its risks. In summary, menopause induces significant cardiovascular changes, primarily due to reduced estrogen levels, leading to unfavorable shifts in lipid profiles, increased blood pressure, and impaired glucose tolerance. These changes result in a higher risk of CVDs, including coronary artery disease, heart failure, and stroke [7, 23, 27].
Comprehensive management strategies are essential to address the interconnected
risks of menopause, estrogen deficiency, and obesity, which contribute to
metabolic syndrome and cardiovascular complications (Fig. 1, Ref. [7, 24, 26, 28]). Menopause leads to a significant drop in estrogen, disrupting
energy balance and contributing to altered body weight, fat distribution, and
increased androgen levels, which elevate obesity risk [6, 7, 23, 25]. Hormonal
changes drive adipocyte hyperplasia and hypertrophy, triggering local
inflammation marked by elevated interleukin-6 (IL-6), tumor necrosis factor-
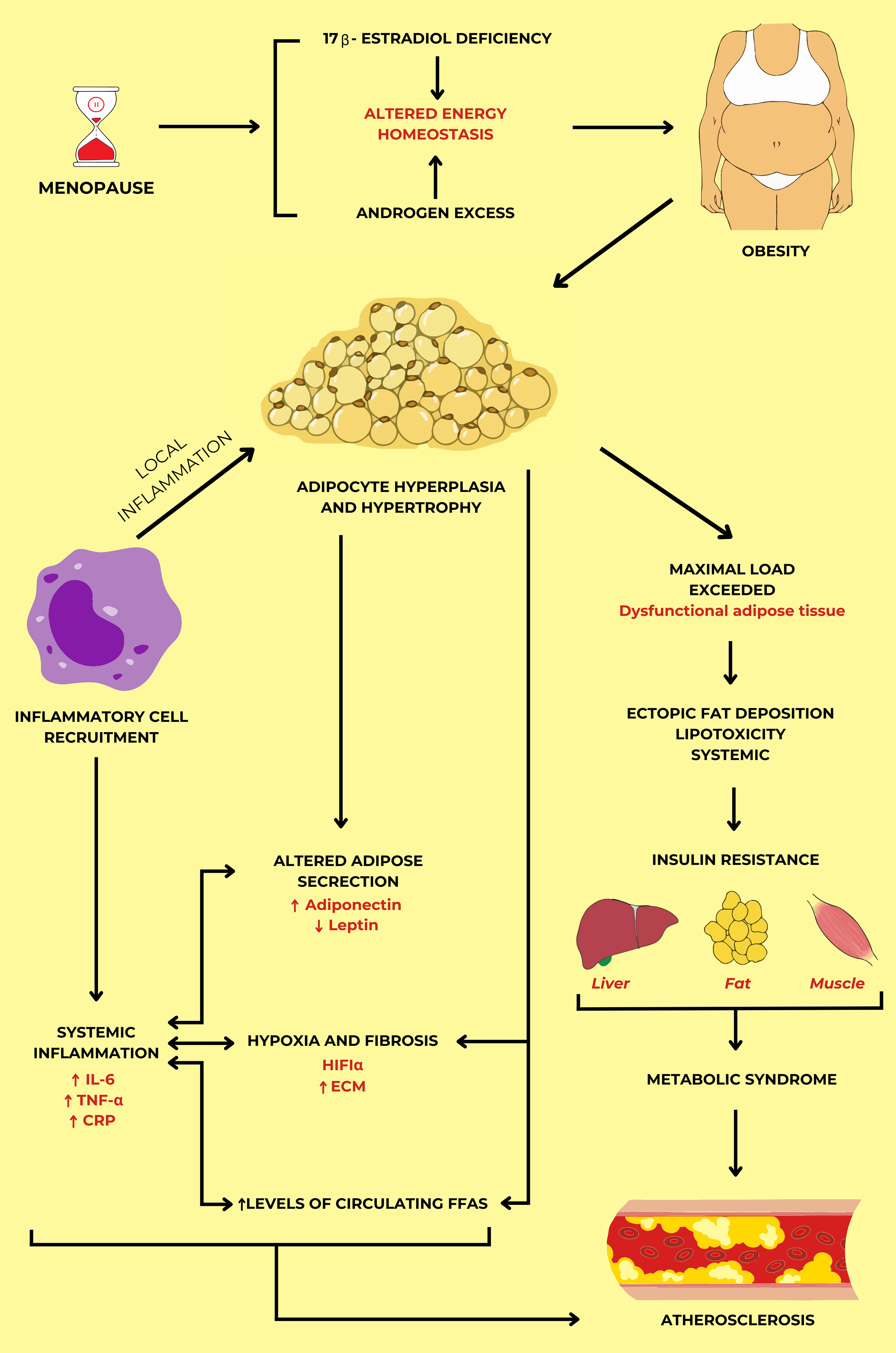 Fig. 1.
Fig. 1.
The mechanism of the development of atherosclerosis in
menopausal women through metabolic syndrome and systemic inflammation. Energy
metabolism changes associated with sex hormone disorders in postmenopausal women
lead to adipose tissue dysfunction in obesity. Due to the insufficient storage
capacity of adipocytes, insulin resistance, and metabolic syndrome appear through
increased lipolysis, hypoxia, and altered adipokine secretion. As a result,
atherosclerosis is established following metabolic syndrome and systemic
inflammation. The figure was drawn by summarizing the findings of references [7, 24, 26, 28] with permission under The Creative Commons Attribution 4.0 Licenses
and Attribution Non-Commercial License. Abbreviations: IL-6, interleukin-6;
TNF
MHT is commonly administered to alleviate menopausal symptoms, such as hot flashes and vaginal dryness, but its effects on metabolic syndrome and CVDs in postmenopausal women are complex and multifaceted. The clinical evidence regarding MHT’s role in managing metabolic syndrome is mixed. On one hand, some studies suggest that MHT, particularly when started around the time of menopause, can improve lipid profiles by increasing HDL cholesterol, decreasing LDL cholesterol, and enhancing insulin sensitivity, which may mitigate the components of metabolic syndrome [23, 24, 25, 31, 32]. Estrogen, the primary hormone used in MHT, has been shown to exert favorable effects on vascular function and glucose metabolism. Conversely, other research has raised concerns about the safety of MHT, particularly regarding its impact on cardiovascular health. Large-scale trials like that by the WHI have revealed that certain forms of MHT may be associated with an increased risk of adverse cardiovascular events, including MI, stroke, and VTE [33, 34, 35]. These risks appear to vary depending on the type of hormone used (estrogen alone vs. estrogen-progestin combinations), the dose, and the timing of initiation relative to the onset of menopause. For example, estrogen-progesterone combinations have been linked with a higher risk of thrombotic events compared to estrogen alone [1, 10, 13, 14]. The timing hypothesis suggests that initiating MHT close to the onset of menopause may offer cardiovascular benefits, whereas starting it later in life could pose increased risks. This has led to the recommendation that MHT should be individualized, taking into account the woman’s overall health, risk factors for CVDs, and the severity of menopausal symptoms [5, 8, 9]. In summary, while MHT might provide benefits in managing metabolic syndrome and improving certain cardiovascular risk factors, its use must be carefully evaluated on a case-by-case basis. Ongoing research and clinical trials aim to clarify the nuanced effects of MHT, refine treatment guidelines, and ultimately ensure that postmenopausal women receive optimal care based on their specific health needs and risk profiles.
The complex network of physiological, neuroendocrine, and metabolic processes
that govern energy balance, body weight regulation, and the role of the
hypothalamus as a central regulator is illustrated in Fig. 2 (Ref. [25]). At the
heart of this system is the hypothalamus, a critical brain region that integrates
and processes a variety of signals from different parts of the body to maintain
energy homeostasis. The hypothalamus receives metabolic signals, such as leptin
and insulin, which are key hormones involved in appetite regulation and energy
expenditure. Leptin, secreted by adipose tissue, provides the brain with feedback
on the body’s energy reserves, while insulin, produced by the pancreas,
influences food intake and energy storage [8, 23, 24, 26]. Fig. 2 also highlights
the role of gastrointestinal satiety signals, including cholecystokinin and
glucagon-like peptide-1, which are released from the stomach and small intestine
in response to food intake [22, 23, 24, 25]. These hormones communicate with the
hypothalamus via the vagal afferent neurons and the paracrine pathway, signaling
fullness and reducing food consumption. The nodose ganglion, which contains the
cell bodies of these afferent neurons, acts as a relay station, transmitting
information from the gut to the brain. The interaction between these signals and
the hypothalamus is essential for regulating appetite and preventing overeating.
Moreover, Fig. 2 illustrates the influence of 17
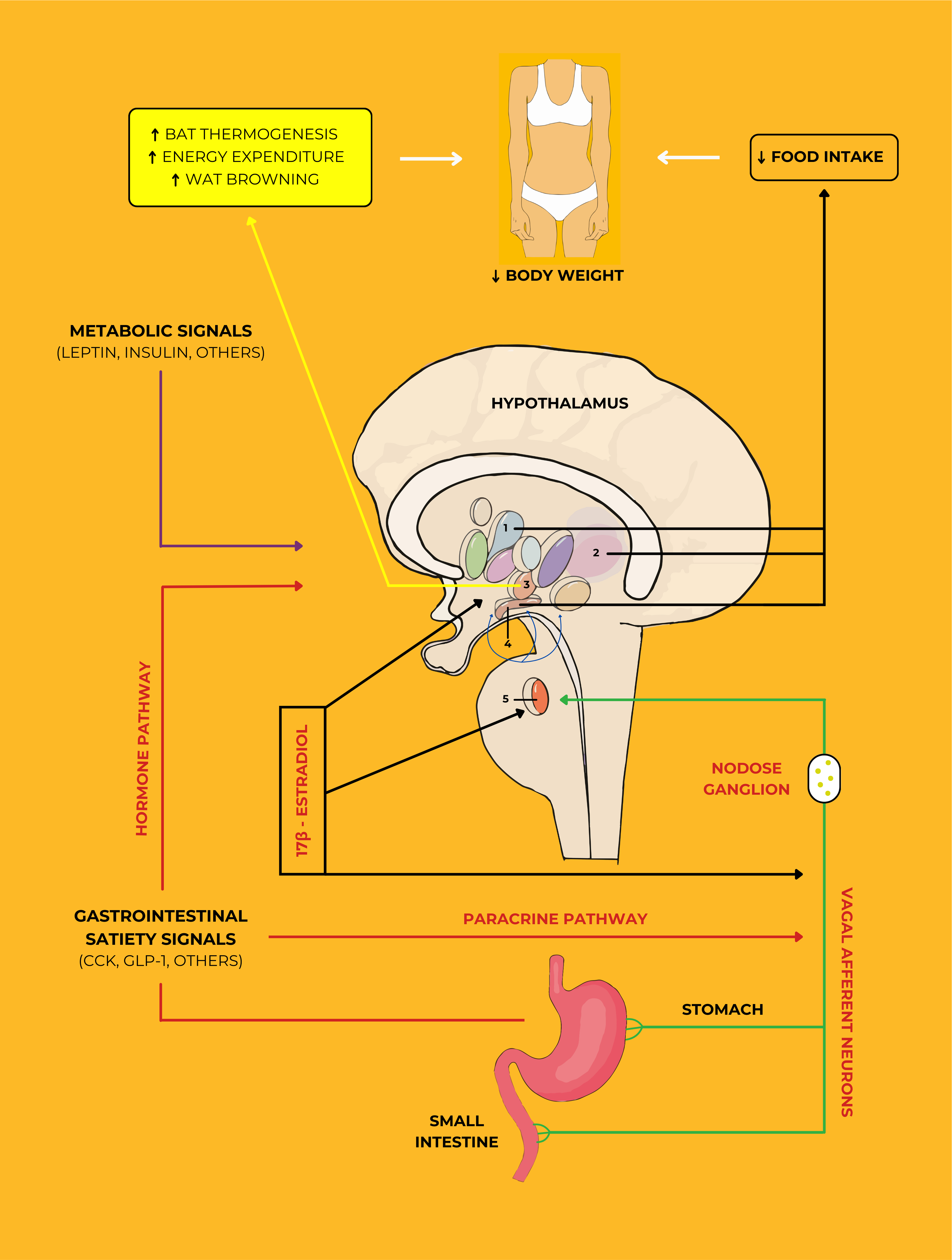 Fig. 2.
Fig. 2.
The essential role of estrogen and meal-related gastrointestinal
signals in reducing body weight. The VANs are activated through a
paracrine-neuronal pathway and then stimulate secondary neurons located in the
NTS (shown in nucleus 5). These signals induce a variety of neurons located in
different nuclei that control feeding behavior in the hypothalamus. While
circulating estrogens respond to these signals by activating on all levels of the
VANs, NTS, and the hypothalamic nuclei such as PVH, LH, VMH, and ARC (shown in
nuclei 1, 2, 3, and 4, respectively), gastrointestinal satiety signals through a
hormonal pathway, as well as metabolic signals, also act upon these nuclei.
Remarkably, estrogens can reduce the levels of AMPK in the VMH. Finally, estrogen
activity through the VMH-SNS-BAT pathway and activated hypothalamic nuclei
contributes to an increase in BAT thermogenesis, energy expenditure, WAT
browning, and a decrease in food intake. This figure was adapted from Vigil
et al. [25] with permission under The Creative Commons Attribution
License. Abbreviations: VANs, vagal afferent neurons; NTS, nucleus of the
solitary tract; PVH, paraventricular hypothalamus; LH, lateral hypothalamus; VMH,
ventromedial hypothalamus; SNS, sympathetic nervous system; ARC, arcuate nucleus;
AMPK, adenosine monophosphate-activated protein kinase; BAT, brown adipose
tissue; WAT, white adipose tissue; CCK, cholecystokinin; GLP-1, glucagon-like
peptide-1;
Fig. 3 (Ref. [38]) depicts the differential effects of MHT on vascular
health during the progression from early to established atherosclerosis in
detail. Fig. 3A focuses on early atherogenesis, wherein the endothelium, the
thin layer of cells lining the blood vessels, plays a vital role in maintaining
vascular homeostasis and preventing the onset of atherosclerosis. In this early
stage, nitric oxide (NO) is a key mediator of vascular health, promoting
vasodilation, which helps maintain open and flexible blood vessels, thereby
reducing blood pressure and improving blood flow. NO also plays a critical role
in reducing inflammatory activation [8, 9, 19, 35, 39]. Inflammation is a driving
factor in atherosclerosis, and by inhibiting inflammatory pathways, NO reduces
the adhesion and migration of leukocytes (white blood cells) to endothelial
cells. This is important because leukocyte adhesion is among the first steps in
atherosclerotic plaque development. NO further contributes to decreasing lesion
progression by limiting the activation of platelets, which are involved in blood
clot formation, and reducing the adhesion of monocytes to the endothelium. Once
adhered, monocytes can migrate into the vessel wall and differentiate into
macrophages, which engulf oxidized LDL particles, leading to foam cell formation,
which characterizes early atherosclerotic lesions [35]. MHT, when administered
during this early stage, can positively modulate these processes. It reduces
levels of endothelin and cyclooxygenase-2, which are associated with
vasoconstriction and inflammation, respectively [40]. It also decreases the
expression of cell adhesion molecules (CAMs) on the endothelial surface, which,
in turn, leads to a reduction in macrophage accumulation and the levels of
pro-inflammatory cytokines such as monocyte chemoattractant protein-1 and
TNF-
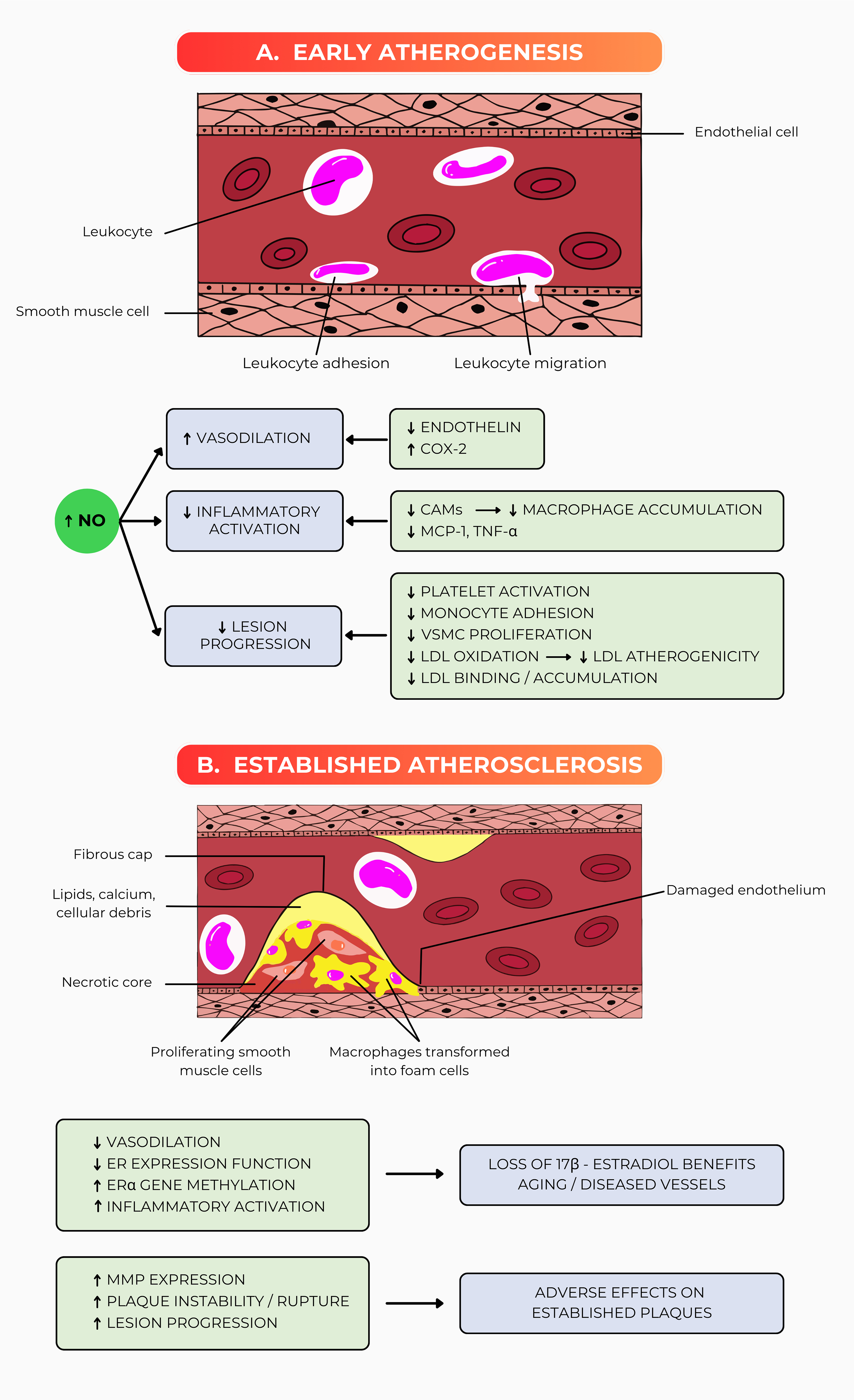 Fig. 3.
Fig. 3.
The benefits of MHT in early atherosclerosis (A) and
its altered biology in established atherosclerosis (B). The endothelium’s
vascular dysfunction is improved by starting MHT early. The delayed onset of MHT
in established atherosclerosis is related to decreased vascular function, which
induces the vulnerability of the vascular wall to inflammatory disturbances. This
figure was adapted from an initiative review [38]
with permission from The American Association for the Advancement of Science.
Abbreviations: COX-2, cyclooxygenase-2; CAMs, cell adhesion molecules; MCP-1,
monocyte chemoattractant protein-1; TNF-
In contrast, Fig. 3B illustrates the scenario in established atherosclerosis,
where the disease has progressed, and protective endothelial functions have
largely been lost. The endothelial cells are now damaged, leading to the
development of a fibrous cap that covers a necrotic core composed of lipids,
calcium, cellular debris, and proliferating smooth muscle cells [6, 26]. These
structural changes make the plaque more prone to rupture, which can precipitate a
heart attack or stroke. In this advanced stage, the benefits of MHT are
diminished, and the therapy may even exacerbate the condition. MHT is associated
with reduced vasodilation, partly due to the decreased expression and function of
estrogen receptors (ERs) on vascular cells, which play a critical role in
mediating the protective effects of estrogen. Increased methylation of the
ER
MHT plays a significant role in managing various aspects of postmenopausal
women’s health, particularly cardiovascular health and metabolic syndrome.
Clinical trial results allow for several conclusions about MHT’s efficacy in
addressing these conditions [13]. With global longevity increasing, women
aged 50 years and older are projected to number 1.6 billion by 2050, up from 1
billion in 2020. Natural menopause occurs at a mean age of approximately 49
years, with vasomotor symptoms like hot flashes and night sweats affecting around
75% of perimenopausal women [45]. These symptoms can persist for a decade or
longer, significantly impacting women’s personal, social, and work lives [13].
Furthermore, up to 84% of postmenopausal women experience genitourinary
symptoms, including vulvovaginal atrophy and incontinence, underscoring the
burden of menopause that MHT aims to alleviate [46]. Clinical guidelines from
various medical societies advocate for MHT to effectively manage these symptoms.
Despite ongoing debates about its broader health effects, the consensus is that
MHT is the most effective treatment available for menopausal symptoms. However,
these guidelines often lack consistency regarding outcomes such as CHD and
all-cause mortality, highlighting the need for further research to evaluate MHT’s
health impacts [1]. For instance, the Heart and Estrogen/Progestin Replacement
Study, one of the first randomized trials of estrogen-progestin therapy for the
secondary prevention of CHD, found no overall cardiovascular benefit and noted an
increase in early CHD events with hormone therapy use, raising concerns about the
timing and appropriateness of prescribing MHT [47]. The landmark WHI trial
further clarified the risks associated with MHT. Enrolling women without CVDs
between the ages of 50 and 79 years, the WHI is the largest randomized
placebo-controlled trial designed to evaluate systemic hormone therapy. Its
initial findings indicated increased risks of CHD, stroke, and VTE among
participants taking MHT compared to those on placebo. However, age-stratified
analyses revealed that younger women (ages 50–59 years) and those who initiated
therapy within a decade of menopause experienced lower absolute risks of adverse
events, supporting the timing hypothesis that MHT’s cardiovascular risks are
influenced by when therapy is initiated relative to menopause [10]. Regarding
long-term use, the Nurses’ Health Study found a significant 41% increase in
breast cancer risk among women over 50 years who used estrogen alone for more
than 20 years, with a 77% increase among lean women [48]. These findings
highlight the complex relationship between MHT and breast cancer risk,
necessitating careful monitoring and individualized treatment approaches.
Research from the Nurses’ Health Study also indicated that women who began MHT
within 4 years of menopause had a significantly reduced risk of CHD, with risk
ratios of 0.66 for estrogen alone and 0.72 for estrogen combined with progestin
[10]. Another study assessed the effects of two hormone replacement regimens in
postmenopausal women, comparing conjugated estrogens with 17
Table 1 (Ref. [5, 6, 9, 10, 18, 23]) shows that estrogen-only therapy has notable positive effects on cardiovascular health, improves lipid profiles, enhances insulin sensitivity, and provides overall relief from menopausal symptoms. In contrast, the estrogen-progesterone combination is effective for symptom relief and supports cardiovascular health, although its effects on lipid profiles may be less pronounced. In conclusion, while MHT offers valuable relief from menopausal symptoms and supports cardiovascular health when tailored appropriately, individual health profiles and initiation timing must be considered carefully. The above findings collectively highlight the need for personalized treatment strategies that consider the timing of initiation, type of therapy, appropriate dosing, and route of administration to optimize outcomes and minimize risks. Ongoing research remains essential for refining guidelines and enhancing the safety and efficacy of MHT for postmenopausal women, ultimately contributing to better health outcomes in this population.
| Parameter | Estrogen-only therapy | Estrogen-progesterone combination |
| Timing of initiation | Best benefits when started around menopause (within 5 years) | Optimal when started within 5 years of menopause, particularly effective for women under 60 years |
| Cardiovascular health | Improved lipid profiles (↑HDL, ↓LDL) | Positive cardiovascular effects when initiated early, especially under 60 years |
| Lipid profile effects | Significant improvements (e.g., 10% ↑HDL, 15% ↓LDL) | Beneficial changes in lipid profiles, but effects may be less pronounced |
| Insulin sensitivity | Enhanced insulin sensitivity | Potential improvements, though variable and not as pronounced |
| Menopausal symptom relief | Effect relief of hot flashes and night sweats | Effective relief of hot flashes, night sweats, and mood swings |
| Mood and sleep | Improvements in mood and sleep quality | Benefits in mood stabilization and improved sleep quality |
| Endothelial function | Enhanced endothelial function | Some improvement is beneficial when started early |
HDL, high-density lipoprotein; LDL, low-density lipoprotein;
Fig. 4 (Ref. [11, 37, 42, 43]) depicts the signaling pathways and cellular
effects mediated by the estrogen hormone 17
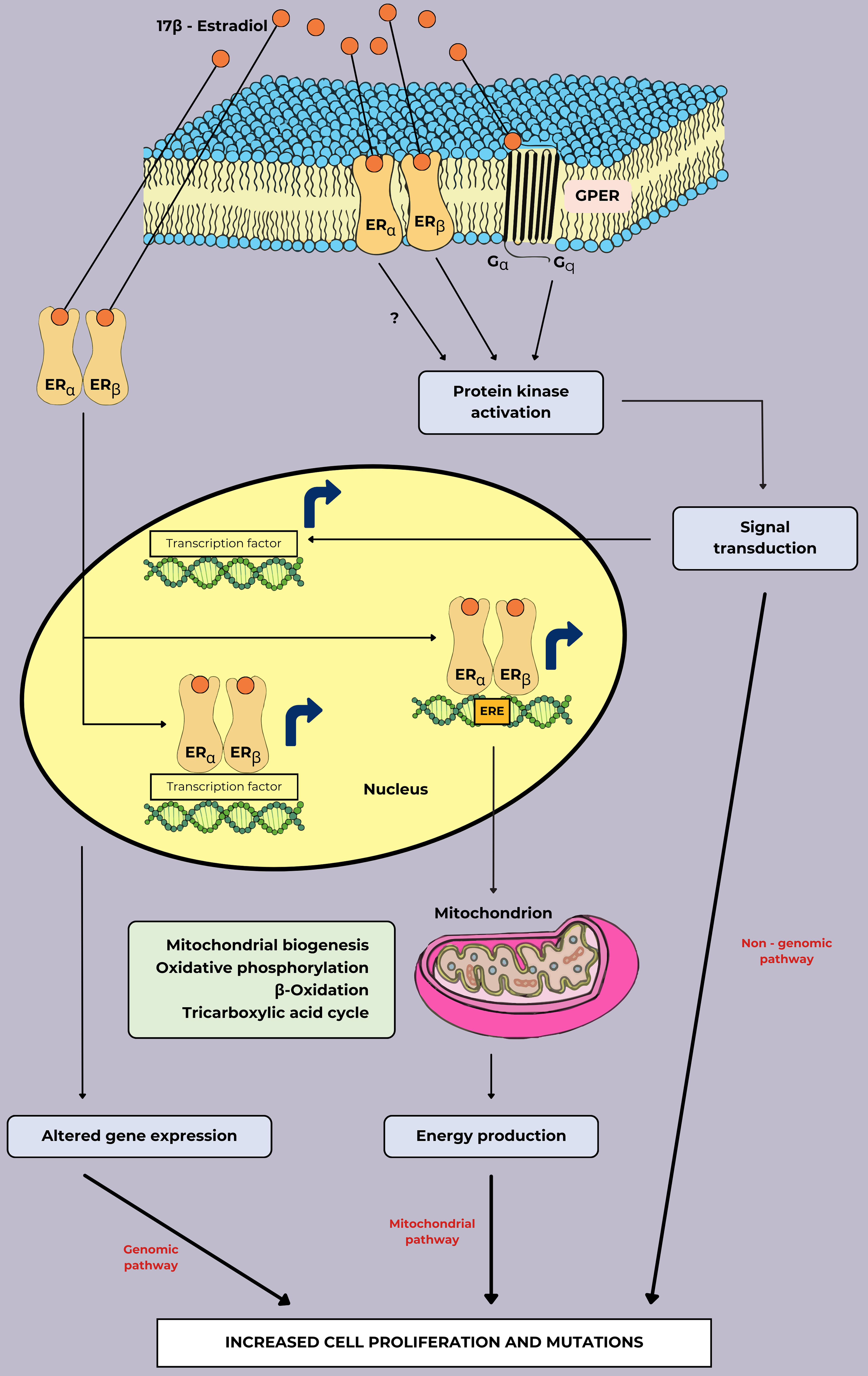 Fig. 4.
Fig. 4.
The signaling pathways of cell proliferation and mutations in breast cancer associated with estrogen treatment. In the genomic pathway, the formation of estrogens and their receptors can induce altered genes that are primarily involved in cell cycle progression, energy metabolism, and survival mechanisms expression by directly binding ERE and transcription factors. In the mitochondrial pathway, essential genes for mitochondrial function are directly activated by the binding of estrogen and ER to ERE. In the non-genomic pathway, estrogen binding to GPER or plasma membrane localization of ER can establish rapid cellular effects by activating several kinase cascades. This figure was adapted from the references [11, 37, 42, 43] with permission under The Creative Commons Attribution 4.0 Licenses. Abbreviations: ER, estrogen receptor; GPER, G protein-coupled estrogen receptor; ERE, estrogen response element.
Fig. 5 (Ref. [53, 54]) depicts 17
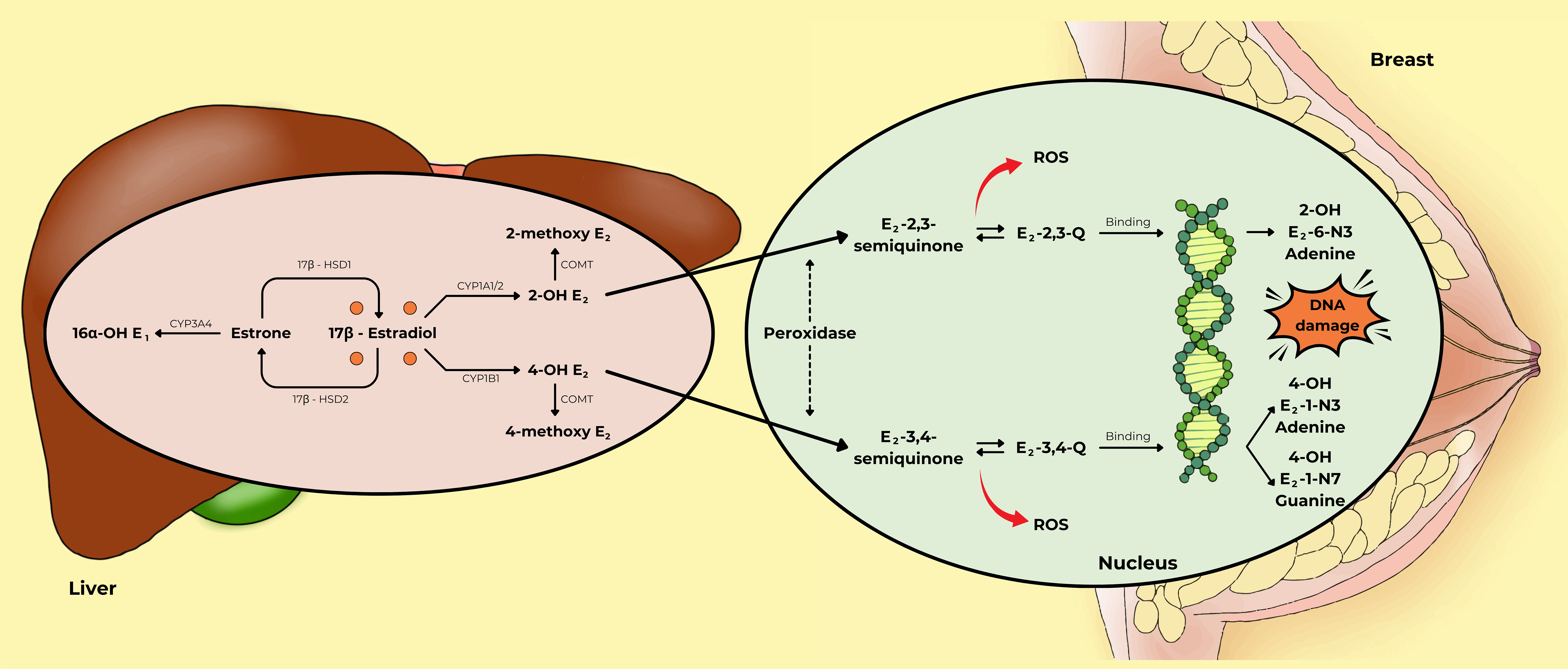 Fig. 5.
Fig. 5.
The metabolism of estrogen plays an important role in
the genotoxic pathway of breast cancer development. In the liver, there are
three main metabolic pathways for estrogen: the 2-hydroxylation pathway, the
4-hydroxylation pathway, and the 16-hydroxylation pathway. In the nucleus, 2-OH
E2 and 4-OH E2 are converted into E2-2,3-Q and E2-3,4-Q by
peroxidase activity. These compounds can bind to DNA to form DNA adducts, such as
2-OH E2-6-N3 adenine, 4-OH E2-1-N3 adenine, and 4-OH E2-1-N7
guanine, which may cause DNA damage in breast cells. This figure was adapted from
the references [53, 54] with permission under The Creative Commons Attribution 4.0
Licenses. Abbreviations: 16
Fig. 6 (Ref. [44, 96, 97, 98]) illustrates the effects of
17
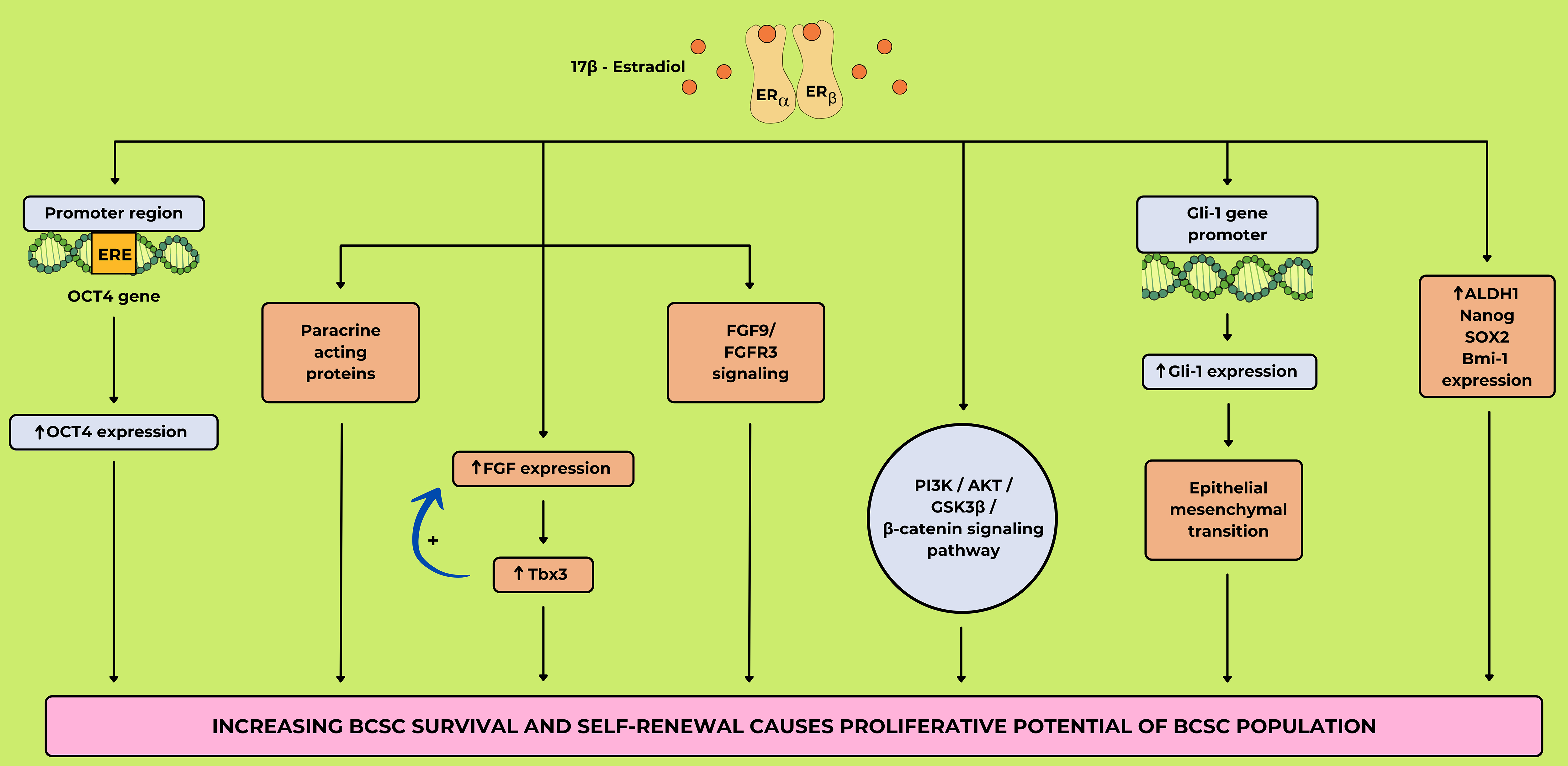 Fig. 6.
Fig. 6.
The mechanism of breast cancer stem cell (BCSC)
expansion through17
The WHI trial, with two arms, generated significant evidence regarding the breast cancer risk associated with MHT. The first arm, focusing on estrogen-plus-progestin therapy, included 16,608 postmenopausal women with an intact uterus who received either 0.625 mg of conjugated equine estrogens (CEE) plus 2.5 mg of MPA daily or a placebo. After a mean follow-up of 5.2 years, the hazard ratio (HR) for invasive breast cancer was 1.26 (95% confidence interval (95% CI), 1.00–1.59), leading to the premature termination of this arm due to the elevated breast cancer risk. The second arm, involving 10,739 postmenopausal women with a prior hysterectomy, investigated the effects of daily CEE alone versus a placebo. After an average follow-up of 7.1 years, the HR for invasive breast cancer was 0.77 (95% CI, 0.59–1.01), suggesting a potential, though statistically non-significant, decrease in breast cancer risk [114, 115]. The Million Women Study, a large observational study in the United Kingdom involving 1,084,110 postmenopausal women aged 50–64 years, found that current users of MHT had a relative risk (RR) of 1.66 (95% CI, 1.58–1.75) for developing breast cancer compared to never-users. The risk was notably higher for those using combined estrogen-progestin therapy (RR: 2.00; 95% CI, 1.88–2.12) than for those using estrogen-only therapy (RR: 1.30; 95% CI, 1.21–1.40) and increased with the duration of use [116]. The Collaborative Group on Hormonal Factors in Breast Cancer (2019) analyzed data from 58 studies, including 108,647 postmenopausal women who developed breast cancer. They found that all types of MHT except vaginal estrogens were associated with an increased risk of breast cancer. Specifically, for women using estrogen-progestin therapy for 5–14 years, the RR was 2.08 (95% CI, 2.02–2.15), while for those using estrogen-only therapy for the same duration, the RR was 1.33 (95% CI, 1.28–1.37) [117]. In a Cochrane Review by Marjoribanks et al. (2017) [118], 22 studies involving 43,637 women were analyzed. The review found that combined continuous MHT was associated with an increased risk of breast cancer, with an RR of 1.26 (95% CI, 1.06–1.51). The absolute risk increase was calculated to be nine additional cases per 1000 women over 5.6 years [118].
MHT remains a critical therapeutic option for menopausal symptoms, but its association with breast cancer risk is a subject of intense study and debate. Varying results from major clinical trials, including the WHI trial and the Million Women Study, underscore the complexity of balancing symptom relief with long-term health outcomes. This discussion focuses on the timing, dose, duration, and types of MHT, followed by insights into future research directions to better understand the risks associated with different MHT regimens. First, the timing of MHT initiation plays a significant role in determining its associated breast cancer risk. Evidence suggests that starting MHT close to the onset of menopause may reduce certain cardiovascular risks while minimizing adverse effects on breast tissue. On the other hand, women who start combined estrogen-progestin therapy shortly after menopause appear to have a slightly higher risk of breast cancer compared to those who initiate therapy later in life, especially after 10 years post-menopause [118]. In addition, according to the British Menopause Society, early initiation of MHT is advised when menopausal symptoms are severe and quality of life is significantly affected. Second, the dose and duration of treatment are emphasized. The Cochrane database has identified a dose-response relationship wherein the lowest effective dose of MHT is given for the shortest possible duration to manage symptoms [118]. In contrast, estrogen-only MHT has a different risk profile. For women who have undergone a hysterectomy, estrogen-only therapy does not seem to significantly elevate breast cancer risk and may even have a protective effect over extended periods of use [115, 119]. Third, the type of MHT regimen is critical in breast cancer risk management. Combined MHT, which includes both estrogen and progestin, is associated with a higher risk of breast cancer than estrogen-only MHT. This elevated risk is particularly pronounced with continuous combined regimens where both hormones are taken daily [114]. For women with a history of breast cancer, systemic MHT is generally contraindicated due to the potential for recurrence, particularly with estrogen-progestin regimens. In such cases, non-hormonal therapies or localized treatments, such as vaginal estrogen for genitourinary symptoms, are often recommended [120, 121, 122]. Given the complexity of individual risk factors and varying responses to therapy, MHT should be personalized. For women at high risk of breast cancer, either due to family history or genetic predisposition, non-hormonal alternatives are often recommended. The risk of breast cancer should also be considered in young women. While the relationship between MHT and breast cancer is now well-established, significant knowledge gaps remain, particularly regarding the long-term effects of newer MHT formulations, such as transdermal patches or bioidentical hormones. Future studies should focus on the comparative risks of different delivery methods, as transdermal estrogen may have a more favorable risk profile than oral preparations due to its lower impact on liver metabolism and clotting factors. Another area requiring more research is the impact of lifestyle factors on MHT-related breast cancer risk. While existing studies have adjusted for obesity, smoking, and alcohol consumption, more granular data are needed to understand how these factors interact with MHT to modulate breast cancer risk. In conclusion, MHT remains a valuable tool for managing menopausal symptoms, but it must be prescribed judiciously, particularly in women at elevated risk of breast cancer. Current evidence supports the use of the lowest effective dose for the shortest duration necessary, emphasizing personalized therapy based on individual risk factors. Future research will help further refine these recommendations and potentially expand the options available to young as well as post-menopausal women who need symptom relief while minimizing breast cancer risk.
Estrogen-only therapy: recent analyses suggest that estrogen-only therapy may be associated with a reduced risk of breast cancer, particularly when initiated early in the menopausal transition. A meta-analysis of randomized controlled trials, including the WHI study, found that women who took estrogen alone had a 35%–37% lower risk of developing breast cancer compared to those who did not receive hormone therapy [123]. Estrogen-only therapy has also been shown to improve cardiovascular outcomes, particularly in women without a uterus. However, it does carry an increased risk of stroke and VTE [124].
Estrogen-progesterone therapy: for women with an intact uterus, estrogen-progesterone therapy is necessary to protect the endometrium from hyperplasia. However, progesterone, particularly synthetic forms like medroxyprogesterone acetate, has been associated with an increased risk of breast cancer [123]. In contrast, micronized progesterone, a more natural form, has not been linked to increased thrombotic risk or higher breast cancer incidence. This combination therapy may provide fewer cardiovascular benefits than estrogen alone, as progesterone has been shown to negatively impact lipid profiles and may increase thrombotic risk [124].
Women at high risk of blood clots: this population includes those with a history of deep vein thrombosis, pulmonary embolism, or inherited blood clotting disorders (e.g., Factor V Leiden mutation). For this group, transdermal estrogen is preferred due to a lower risk of VTE compared to oral forms, as it bypasses the liver’s first-pass metabolism [124].
Women with cardiovascular risk factors: this group comprises individuals with conditions such as hypertension, hyperlipidemia, or a family history of CVD. MHT may have cardiovascular benefits if started early in menopause (before age 60 years or within 10 years of onset). Estrogen-alone therapy, when appropriate, could lead to better cardiovascular outcomes, but decisions should be individualized based on comprehensive risk assessments [124].
Obese women or those with metabolic syndrome: studies showed that obese female students faced menorrhagia and estradiol may deal with body weight regulation [25, 125]. Women with obesity, insulin resistance, or metabolic syndrome often have a higher risk of complications from MHT. For them, low-dose or transdermal estrogen might be safer options, as these can mitigate the metabolic impact [124].
Women with a history of breast cancer or high breast cancer risk: systemic MHT is generally contraindicated in women with a personal history of breast cancer due to an increased risk of recurrence. However, localized vaginal estrogen may be considered for severe genitourinary symptoms, as it has a minimal systemic effect [123].
Women with persistent, severe menopausal symptoms: for women whose quality of life is significantly affected by menopausal symptoms and who do not respond well to non-hormonal treatments, MHT may be considered after carefully weighing the risks and benefits and closely monitoring their health outcomes.
Overall, these findings emphasize the need for individualized treatment based on a patient’s health history, age, and specific risks. Estrogen-only therapy may be more beneficial for those without a uterus and with lower cardiovascular risks, while estrogen-progesterone therapy remains necessary for those with an intact uterus despite its additional risks. Special consideration is needed for women in high-risk groups, such as those with a history of blood clots, CVD, obesity, or breast cancer. For these populations, transdermal estrogen or low-dose options are often recommended, as they may be safer alternatives with fewer metabolic impacts and thrombotic risks. Personalized care is essential, accounting for individual health factors to optimize the benefits of MHT while minimizing potential risks.
In conclusion, MHT has significant benefits, alleviating menopausal symptoms, improving quality of life, and potentially reducing cardiovascular risks when initiated early in menopause. Timing plays a critical role, as early initiation may help reduce the progression of atherosclerosis, whereas late introduction can pose greater risks. Estrogen-only therapy, typically used in women without a uterus, has been associated with greater cardiovascular benefits and a reduced risk of breast cancer but carries risks of stroke and VTE. In contrast, estrogen-progesterone therapy, required for women with an intact uterus to prevent endometrial hyperplasia, offers symptom relief but is linked to a higher risk of breast cancer and may have diminished cardiovascular benefits. These benefits and risks must be carefully balanced, particularly in older women or those with pre-existing conditions. The relationship between MHT and breast cancer involves multiple mechanisms, including genomic, non-genomic, and genotoxic pathways, as well as the expansion of BCSC. Given the complexity of each patient’s health profile, the personalization of MHT is paramount. Recommendations include using the lowest effective dose for the shortest duration while tailoring therapy to patient-specific risk factors, such as age, time since menopause onset, obesity, smoking, and alcohol use. By adopting a personalized approach, clinicians can optimize MHT’s therapeutic potential while minimizing adverse effects, ensuring that each patient receives the most appropriate and effective care.
TTTT and THN designed the research study. THN was responsible for manuscript writing. The table was conducted by ATMD while the graphic figures were created by TMP, the data from studies were compiled by QTML. ATMD and QTML contributed to preparing draft and editorial revisions. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated suffciently in the work and agreed to be accountable for all aspects of the work.
During the preparation of this work, the authors used ChatGpt-3.5 and QuillBot to check spelling and grammar. After using this tool, the authors reviewed and edited the content as needed and took full responsibility for the content of the publication. The authors independently reviewed and edited the content to ensure its accuracy and quality, taking full responsibility for the final content of the publication.
Not applicable.
We gratefully acknowledge Can Tho University of Medicine and Pharmacy for the time and effort they devoted to the study. Furthermore, we also thank all the editors and peer reviewers for their opinions and suggestions.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.