





 , Ralf Kinscherf 1, Gabriel A. Bonaterra 1
, Ralf Kinscherf 1, Gabriel A. Bonaterra 11 Department of Medical Cell Biology, Institute for Anatomy and Cell Biology, Philipps-University of Marburg, 35037 Marburg, Germany
Abstract
Stress- and inflammation-induced growth differentiation factor-15 (GDF-15) is proposed as a biomarker for mortality and disease progression in patients with atherosclerosis and/or cardiovascular disease (CVD). The development of atherosclerotic lesions depends, among other factors, on inflammatory processes, oxidative stress, and impaired lipid homeostasis. As a consequence, activation and dysfunction of endothelial cells, release of chemokines, growth factors and lipid mediators occur. GDF-15 is suggested as an acute-phase modifier of transforming growth factor (TGF)-ßRII-dependent pro-inflammatory responses leading to rupture of atherosclerotic plaques, although the exact biological function is poorly understood to date. GDF-15 is upregulated in many disease processes, and its effects may be highly context-dependent. To date, it is unclear whether the upregulation of GDF-15 leads to disease progression or provides protection against disease. Concerning CVD, cardiomyocytes are already known to produce and release GDF-15 in response to angiotensin II stimulation, ischemia, and mechanical stretch. Cardiomyocytes, macrophages, vascular smooth muscle cells, endothelial cells, and adipocytes also release GDF-15 in response to oxidative as well as metabolic stress or stimulation with pro-inflammatory cytokines. Given the critically discussed pathophysiological and cellular functions and the important clinical significance of GDF-15 as a biomarker in CVD, we have summarized here the basic research findings on different cell types. In the context of cellular stress and inflammation, we further elucidated the signaling pathway of GDF-15 in coronary artery disease (CAD), the most common CVD in developing and industrial nations.
Keywords
- GDF-15
- inflammation
- stress
- coronary artery disease
Growth differentiation factor-15 (GDF-15), which is identical to macrophage
inhibitory cytokine-1 (MIC-1), prostate-derived factor (PDF), nonsteroidal
anti-inflammatory drug (NSAID)-activated gene-1 (NAG-1), placental bone
morphogenetic protein (PLAB), and placental transforming growth factor (PTGF)
[1, 2, 3, 4, 5, 6], is a divergent member of the transforming growth factor-
Many clinical trials revealed elevated plasma/serum GDF-15 levels in various diseases, thus, indicating that GDF-15 may be considered as a biomarker. These pathophysiological conditions and diseases associated with increased plasma/serum GDF-15 levels include endothelial activation and vascular inflammation, which determine the development and progression of atherosclerosis, cardiovascular disease (CVD) and/or cardiometabolic diseases [16, 17, 18], heart failure [19, 20], lipodystrophy [15, 21], or even cancer [10, 22, 23, 24, 25, 26]. With respect to CVD, macrophages, VSMCs, ECs, adipocytes, and cardiomyocytes produce and release GDF-15 in high concentrations in response to mitochondrial dysfunction, oxidative stress, metabolic stress, and/or through stimulation by pro-inflammatory cytokines [1, 13, 15, 27, 28] (Fig. 1).
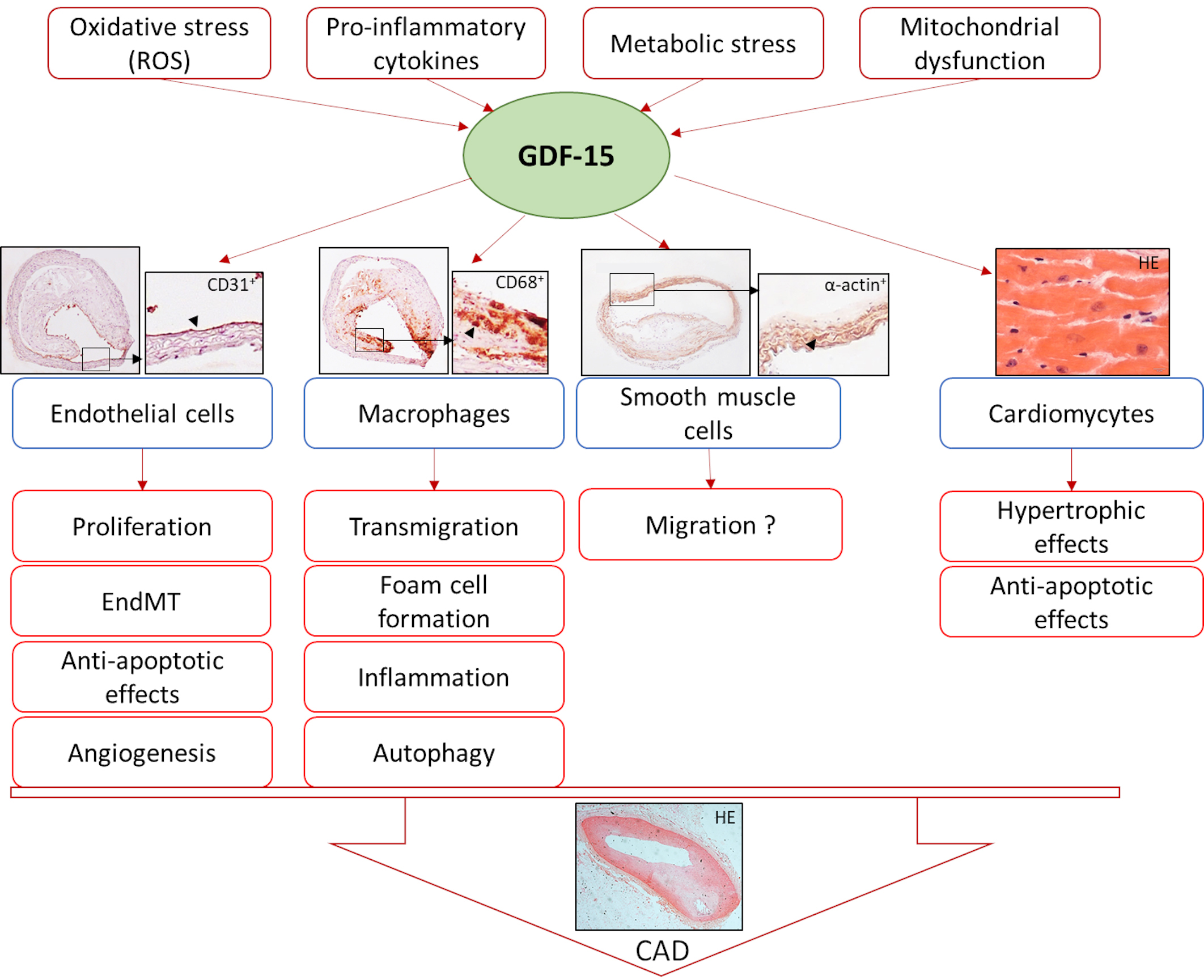 Fig. 1.
Fig. 1.Induction of GDF-15 and its effects on different cell types leading to coronary artery disease (CAD). HE, Hematoxylin-eosin stain.
Previous studies have shown, that the physiological effect of GDF-15 is highly context-dependent and can vary significantly with the stage of disease [29]. Therefore, we would like to use this review to summarize the actual existing research data and focus on the effect of GDF-15 in different cell types with special reference to cellular stress and inflammation to better understand the signaling pathways of GDF-15 in coronary artery disease (CAD).
CAD, also named coronary heart disease (CHD), ischemic heart disease (IHD), or myocardial ischemia is a chronic heart disease caused by atherosclerotic plaques in the coronary arteries leading to more or less coronary stenosis. In this context, several studies have shown that GDF-15 is useful as a consistent biomarker of mortality and CV events in patients with acute coronary syndrome (ACS) [30, 31, 32, 33, 34], acute Heart Failure [35] or stable CAD [34, 36, 37, 38, 39] (Table 1, Ref. [30, 32, 33, 34, 37, 38, 39, 40, 41, 42].
| Study | Population | Median GDF-15 concentration at baseline (ng/L) | GDF-15 assays | Follow up (Median) | Reference |
|---|---|---|---|---|---|
| Gusto-4 | 2081 patients; NSTE-ACS | 1434 (1035–2078) (validation cohorte) | IRMA | 1 year | [30] |
| 1499 (1151 to 2203) (derivation cohort) | |||||
| Assent-2 and assent-plus | 741 patients; STEMI | 1635 (1164–2309) | IRMA | 1 year | [33] |
| Atherogene | 1352 patients with SAP, 877 patients with ACS | SAP: 1128 (850–1553) ACS: 1244 (962 to 1785) | immunoradiometric assay (IRMA) | 3.6 years | [34] |
| Leicester royal infirmary infarct registry | 1142 patients; NSTEMI or STEMI | 1470 (240–31,860) | ELISA (antibodies from R&D) | 1.4 years | [32] |
| Prove it-timi-22 | 3501 patients; NSTE-ACS or STEMI | 1362 (1032–1844) | IRMA | 2 years | [41] |
| Heart and soul | 984 patients; stable CHD | 2166 (1589–3057) | Luminex Sandwich Assay (Alere Diagnostics, San Diego, CA) | 8.9 years | [39] |
| Iabp-shock-2 | 190 patients NSTEMI or STEMI and cardiogenic shock undergoing primary PCI | 7662 | Quantikine ELISA (R&D) | 30 days | [42] |
| Karola | 1029 patients; stable CHD, History of MI or CABG | 1232 (916–1674) | ElectroChemi-Luminescence Immunoassays (Fa. Roche) | 10 years | [38] |
| Plato | 16,876 patients NSTE-ACS or STEMI | 1550 (1145–2219) | ElectroChemi-Luminescence Immunoassays (Fa. Roche) | 1 year | [40] |
| Stability | 14,577 patients; stable CHD | 1253 (915–1827) | ElectroChemi-Luminescence Immunoassays (Fa. Roche) | 3.7 years | [37] |
The GUSTO-4 (Global Utilization of Strategies to Open Occluded Arteries-4) trial demonstrated a strong association between GDF-15 concentration in the blood of patients at hospital admission and all-cause mortality in non-ST-segment-elevation ACS (NSTE-ACS) [30] (Table 1). In the samples of 2081 patients with NSTE-ACS, increasing levels of GDF-15 at admission were positively associated with age, female sex, hypertension, and diabetes [30]. GDF-15 levels were also associated with previous manifestations of heart disease, current angiotensin-converting enzyme inhibitor therapy, and markers of ongoing ischemia and necrosis, myocardial dysfunction, and inflammation [30]. In addition to independent risk indicators such as, age, N-terminal pro-brain natriuretic peptide (NT-proBNP), and myocardial infarction, GDF-15 was the most important predictor of death in this study [30]. By determining 1-year cumulative mortality rates, GDF-15 was one of the best predictors provided prognostic information more than other clinical biomarkers (cardiac troponin-T [cTnT], NT-proBNP, hs- C-reactive protein [CRP], and creatinine clearance) in the comparison [30]. In patient groups with NSTE-ACS or ST-elevation myocardial infarct (STEMI), the independent association of GDF-15 with mortality was reconfirmed [32, 33]. In patients with NSTE-ACS or STEMI the prognostic value of GDF-15 was reassessed, in the Platelet Inhibition and Patient Outcomes Trial (PLATO) [40] (Table 1). Because of the large number of patients, the PLATO biomarker study examined the association between GDF-15 and specific outcome events during follow-up. After adjusting for clinical predictors and biomarkers (hs-cTnT, NTproBNP, hs-CRP, and cystatin C), the study showed that elevated GDF-15 levels were associated with an increased risk of, CV mortality, myocardial infarction, and stroke [40].
The AtheroGene registry enrolled patients with stable angina pectoris (SAP) or
ACS who underwent coronary angiography and had stenosis of
The Heart and Soul Study with 984 patients examined the effects of psychosocial factors on the health status of patients with stable heart failure (Table 1). In this study, GDF-15 was independently associated with fatal and nonfatal CV events, and hospitalization for heart failure in stable CAD during nearly 9 years of follow-up [39]. In addition, this study demonstrated that higher GDF-15 plasma levels belong to a lower left ventricular ejection fraction (LVEF), diastolic dysfunction, greater inducible ischemia, and lower-body exercise output [39]. The Stabilization of Atherosclerotic Plaque by Initiation of Darapladib Therapy trial (STABILITY) evaluated the effectiveness of the inhibitor of lipoprotein-associated phospholipase A2 (Lp-PLA2), Darapladib, compared with placebo during a median follow-up of 3.7 years, assessing the incidence of CV events in 15,828 patients with stable CAD receiving secondary preventive treatment [37] (Table 1). Additionally, blood samples were obtained from patients with stable CAD, demonstrating that higher GDF-15 plasma concentrations at baseline were associated with an increased event rate of the primary composite end point (death from CVD, nonfatal myocardial infarction, or nonfatal stroke) [37]. In multivariable-adjusted analyses, higher GDF-15 plasma concentrations were associated with age and gender [37]. Risk factors like advanced age, male gender, smoking, hypertension, diabetes mellitus, renal dysfunction, poly-vascular disease, hypertriglyceridemia, leucocytosis, and lower concentrations of hemoglobin and HDL-C were related to GDF-15 plasma concentrations [37]. Similarly, increased GDF-15 plasma concentrations correlated with higher concentrations of NT-proBNP, hs-troponin T, and cystatin C [37, 40]. This study proved that in patients with stable CAD, GDF-15 is an independent risk marker associated with CV and non-CV death [37]. The KAROLA cohort is a prospective study of 1204 CAD patients enrolled in a cardiac rehabilitation program after ACS or coronary artery bypass grafting (CABG) surgery [38] (Table 1). The KAROLA study included patients with stable CAD and a follow-up period of 10 years. This study also demonstrated that baseline GDF-15 levels were associated with the occurrence of a subsequent CV event and all-cause of death after adjustment for established CV risk factors [38].
Data from the above-mentioned clinical trials, indicate that the baseline of GDF-15 plasma concentrations and their changes over 12 months provide important prognostic information for identifying patients at high risk of mortality. In reviewing these various clinical studies, GDF-15 (especially its concentration in plasma/blood) may be suggested as a biomarker for CVD and severity. However, it remains unclear, whether the GDF-15 pathway has therapeutic potential.
Chronic vascular inflammation, oxidative stress, and endothelial dysfunction are
hallmarks of the development and progression of atherosclerotic lesions in
coronary arteries resulting in CAD [43, 44, 45]. The imbalance of reactive oxygen
species (ROS) and antioxidant defenses is one of the main causes of endothelial
dysfunction [43]. Increased NADPH oxidase (Nox) activity uncouples endothelial
NO-Synthase (eNOS), increases ROS, and decreases nitric oxide (NO)
bioavailability [46]. NO is a strong vasodilator that also inhibits the
expression of transcription factors such as NF-
| Cell Type | Clinical relevance | Effects | Mechanisms/ Molecules | References |
| HPMEC | PAH | proliferation |
AKT | [52] |
| HUVEC | Tumor-angiogenesis | angiogenesis |
phosphorylation of Rb protein, nuclear translocation of E2F-1, AP-1- and E2F-dependent expression of G1 cyclins via PI3K/AKT, JNK, ERK signaling pathways | [53] |
| HUVEC | Cardiac ischemia | angiogenesis |
p53, HIF-1 |
[59] |
| HUVEC | diabetes mellitus, hyperglycemia | apoptosis |
NF- |
[55] |
| HUVEC | Cardiac disease, cancer | angiogenesis |
CCN2-mediated angiogenesis, |
[58] |
| HUVEC | regenerative medicine of calvarial defect | proliferation |
PI3K/AKT, JNK, ERK signaling pathways | [54] |
| human umbilical vein cell line EA.hy926 | Hepatocellular carcinoma | angiogenesis |
Src, AKT, MAPK-, NF-κB-signaling pathway | [60] |
| HAEC | CVD in women | proliferation |
p53 pathway | [56] |
| Endothelial Colony Forming Cells from adult blood | Senescence | proliferation |
NO |
[57] |
Studies of vascular remodeling in pulmonary arterial hypertension (PAH), which
is characterized by endothelial dysfunction with release of vasoactive mediators,
growth factors, and cytokines [61], show that GDF-15 is increased in PAH lungs,
predominantly located in vascular ECs [52]. PAH is characterized by pulmonary
vascular remodeling, progressive arterial stiffening, increased vascular
resistance, and right ventricular failure. Animal and human studies suggest with
growing evidence that ROS and oxidative stress play a key role in the
pathogenesis of PAH [61, 62]. In vitro analyses of human pulmonary
microvascular endothelial cell (HPMEC) proliferation and apoptosis suggest a role
for GDF-15 in endothelial cell homeostasis in PAH patients [52]. HPMEC showed
marked upregulation of GDF-15 in hypoxia and laminar shear stress [52].
Recombinant (r) GDF-15 protein decreased apoptotic cell death of HPMEC. In
contrast, proliferation was either increased or decreased depending on the
concentration of rGDF-15 protein [52] (Table 2). Further studies showed that
GDF-15 stimulated the proliferation of human umbilical veins endothelial cells (HUVECs) by
upregulating cyclins D1 and E via the phosphoinositide 3-OH kinase (PI3K)/
protein kinase B (AKT) signaling pathway, extracellular signal-regulated kinases
(ERK), and c-Jun N-terminal kinase (JNK)-dependent AP-1 and E2F activation
signaling pathways [53, 54] (Fig. 2). The effect of GDF-15 against apoptotic cell
death might be related to influence on PI3K/AKT/eNOS pathway and
NF-
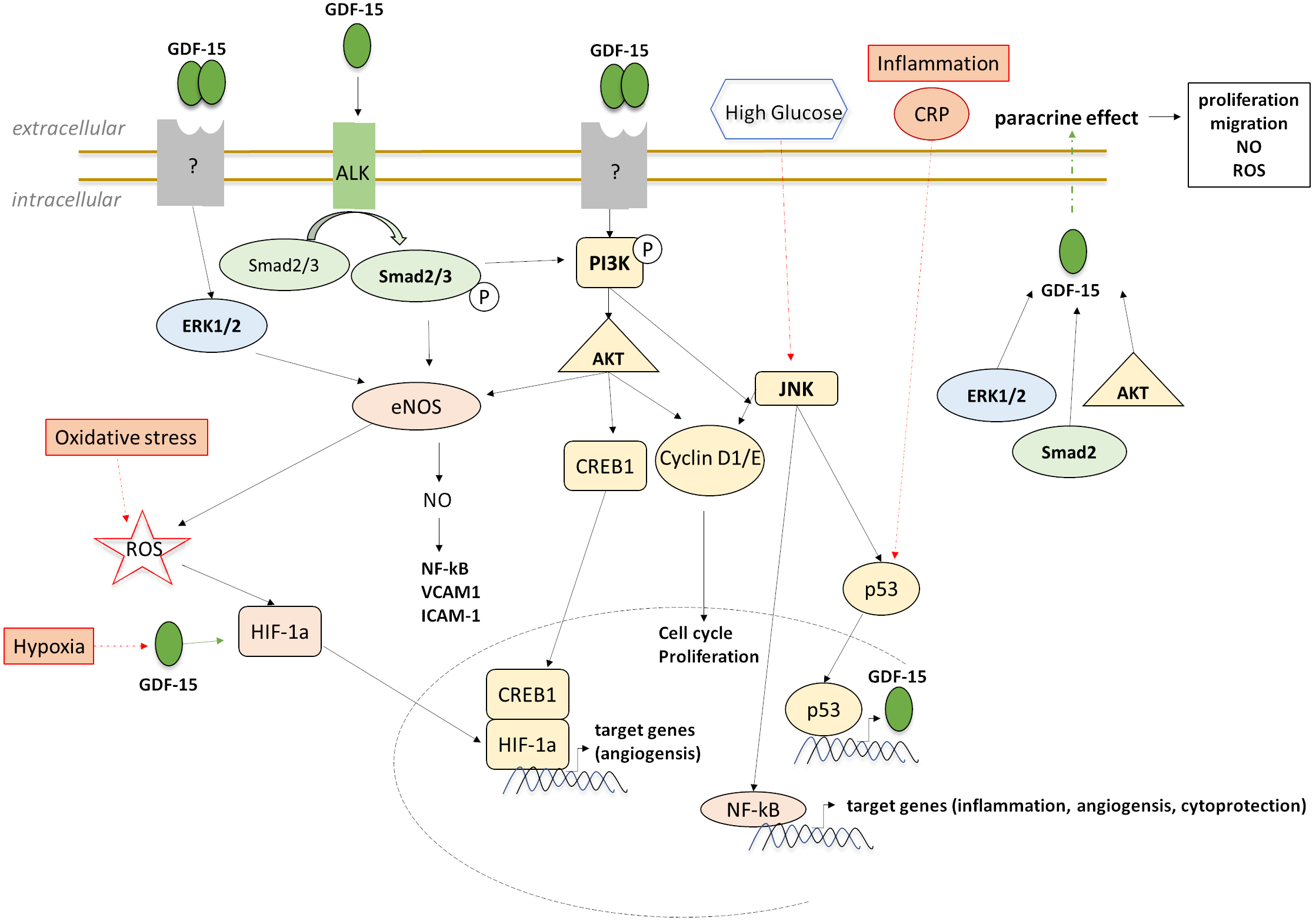 Fig. 2.
Fig. 2.Downstream targeting and signaling of GDF-15 in ECs in experimental stress-induced models.
Cell senescence is a mechanism of aging and plays a vital role in the onset and prognosis of CVD [63]. Increasing evidence shows that cell senescence is indispensable in the formation and development of atherosclerosis [63]. To investigate GDF-15 expression, function and role during cellular senescence, Ha et al. [57] studied endothelial colony-forming cells (AB-ECFCs) as a model for ECs, because cell senescence is mainly involved in vascular stress and loss of endothelial function. They found that AB-ECFCs expressed higher levels of GDF-15 compared with cord blood colony-forming cells (CB-ECFCs) and that GDF-15 expression progressively increased as AB-ECFCs senescent [57]. Previous studies showed that GDF-15 was overexpressed in radiation-induced senescent HAECs [64]. The paracrine action of GDF-15 promotes AB-ECFC proliferation, migration, and NO production through activation of AKT, ERK, and Mothers against decapentaplegic homolog 2 (SMAD2) signaling pathways. It induces ROS production independently of nuclear factor-like 2 (NERF2), the major transcription factor regulating antioxidant response [57] (Fig. 2). Ha et al. [57] interpreted the paracrine effect of GDF-15 by senescent AB-ECFCs on non-senescent AB-ECFCs as a benefit and claimed that GDF-15 might play a beneficial role in a dysfunctional vasculature by limiting endothelial dysfunction associated with vascular stress.
An increase in endothelial permeability and micro vascularization in the plaque
are critical factors in the atherogenesis. Regarding the angiogenic process,
Whitson et al. [58] described that GDF-15 interacts with connective
tissue growth factor 2 (CCN2), inhibits CCN2-mediated angiogenesis, and blocks
CCN2-mediated tube formation in HUVECs. However, in hypoxic HUVECs Song
et al. [59] described that GDF-15 promotes angiogenesis via the
hypoxia-inducible factor 1-alpha (HIF-1
An induction of GDF-15 has been reported and described in numerous diseases, such as CVD, cancers, metabolic disorders, rheumatic diseases and viral infection [49, 65, 66, 67, 68]. The majority of these diseases are associated with inflammation and cellular stress.
TGF-ß family members, including GDF-15, have effects on cell proliferation,
differentiation, apoptosis and inflammation as well as cellular motility and
adhesion [69, 70]. The expression of GDF-15 was examined in the human monocytic
cell lines U937, KG-1 and THP-1 [1, 71], whereby under (oxidative) stress
conditions, such as incubation with trans-retinoic acid (RA) and phorbol 12
myristate 13-acetate (PMA), oxidized low-density lipoprotein (oxLDL),
C6-ceramide, or H
| Cell Type | Clinical relevance | Effects | Mechanisms | References |
| U937, KG-1 | paracrine/autocrine effect | Inflammation |
LPS-induced TNF- |
[1] |
| Human peripheral blood mononuclear cells | atherosclerosis | Inflammation, oxidative stress | GDF-15 |
[13] |
| RAW 264.7, bone marrow-derived macrophages | vascular injury | Chemotaxis |
S/G2 phase arrest, CCR2 | [75] |
| polymorphonuclear leukocyte | myocardial infarction | leukocyte ß |
GTPase Cdc42, Rap1 | [77] |
| GDF-15 |
atherosclerosis | inflammation |
IL-6, Caspase-3 | [49] |
| RAW 264.7, NAG-1 |
obesity, intestinal cancer | Leptin expression |
GDF-15 does not directly inhibit the TLR4/NF |
[73] |
| PMA-differentiated THP-1 macrophages | Foam cells, oxLDL | Cholesterol Efflux |
ABCA1, PI3K/PKC/SP1 pathway | [76] |
| Kupffer cells | acute liver injury | inflammation |
IκB |
[74] |
| CD11b |
Viral and bacterial infection, Sepsis | LPS-responds | GDF-15 |
[72] |
| PMA-differentiated THP-1 | Foam cells, oxLDL | Autophagy |
ATG5, p62-accumulation, LC3II/I | [48, 71] |
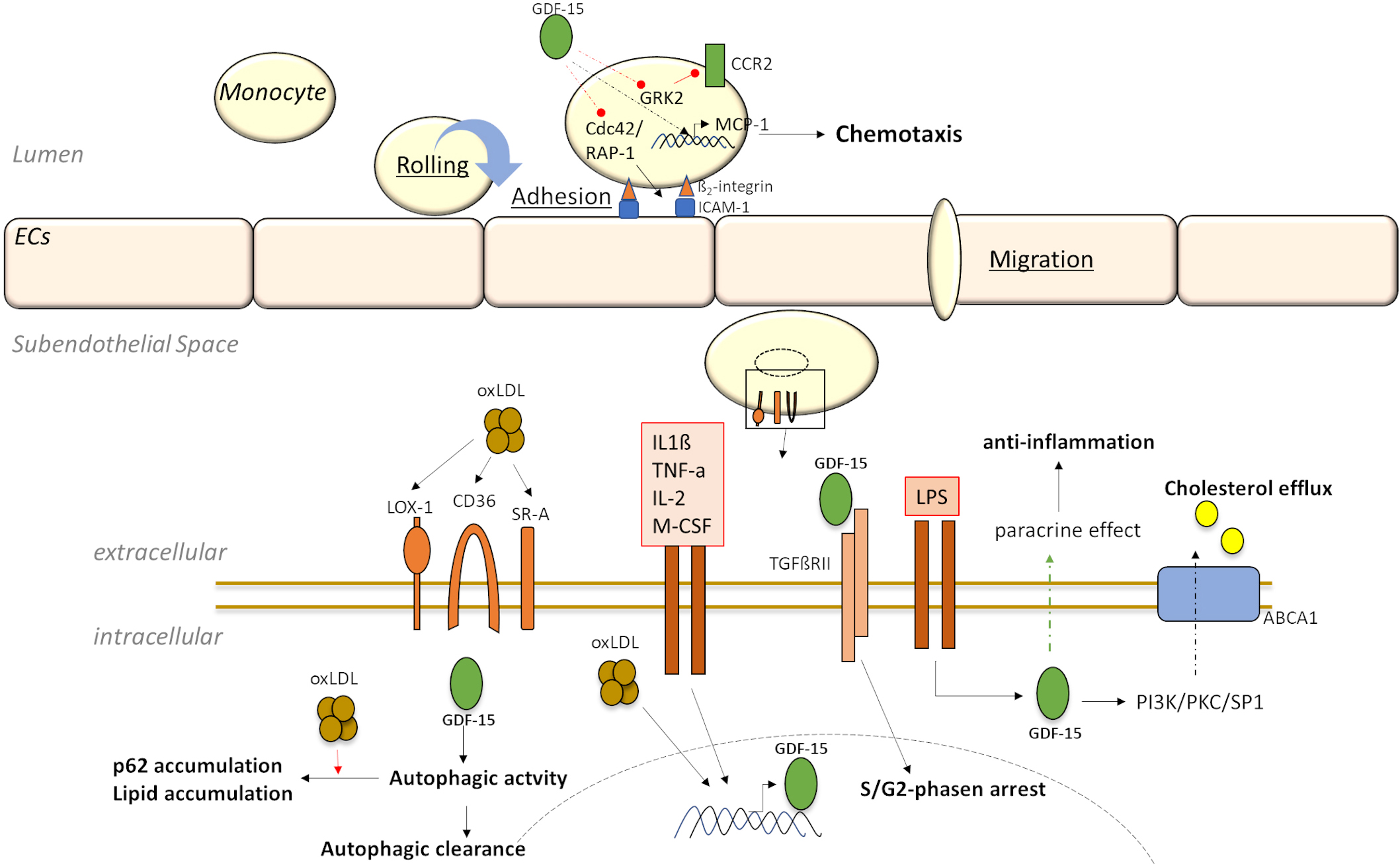 Fig. 3.
Fig. 3.Downstream targeting and signaling of GDF-15 in monocytes/macrophages in experimental stress-induced models.
In relation to atherosclerotic research studies, immunohistochemical analyses of
human atherosclerotic carotid arteries have demonstrated colocalization of GDF-15
with oxLDL, CD68 immunoreactive cells and apoptosis-relevant proteins [13, 75]. In
this context, GDF-15 was up-regulated in murine atherosclerotic lesions during
disease progression in a pattern similar to CD68
In this context, in vitro data have proved that GDF-15 deficiency leads to a decreased mRNA expression of apoptosis- or inflammation-relevant cytokines in cultured peritoneal macrophage of mice [49]. Additionally, data from human PMA-differentiated THP-1 macrophages have suggested that GDF-15 is involved in the regulation of lipid homeostasis by regulating autophagic processes [48, 71](Fig. 3; Table 3). Studies using small interfering RNA against GDF-15 (siGDF-15) and recombinant GDF-15 have demonstrated that GDF-15 directly affects autophagic activity in macrophages without affecting lysosomal activity [48, 71]. Also, in combination with oxLDL, GDF-15 affects autophagic processes with consequences for lipid homeostasis in human macrophages [71] (Fig. 3; Table 3), indicating its emerging important pathophysiological role in the development and progression of atherosclerotic plaques. In the context of foam cell formation, another study using THP-1 macrophages has demonstrated that GDF-15 might be a potential target to prevent foam cell formation via the PI3K/PKC/SP1 pathway and promote cholesterol efflux [76] (Fig. 3; Table 3). Therefore, GDF-15 has been shown to regulate apoptosis, autophagy and inflammatory processes of macrophages and is involved in configuring atherosclerotic lesion development.
In terms of the selectin-mediated leukocyte capturing and rolling, followed by
the actual transmigration through the endothelium, resulting in chemokine-induced
leukocyte arrest [83, 84], GDF-15 is essential to prevent the excessive
chemokine-activated leukocyte arrest and transmigration through the endothelium
[77]. Additionally, GDF-15 is an inhibitor of leukocyte ß
In the context of CAD, smooth muscle cells (SMCs) play a key role in the
stability and progression of atherosclerotic plaques. In addition to macrophages
and ECs, as cellular sources of GDF-15 production, VSMCs also secrete GDF-15 in
response to metabolic and/or oxidative stress or stimulation by pro-inflammatory
cytokines [28]. In studies concerning atherosclerotic plaques of the pulmonary
trunk of GDF-15 deficient ApoE
After a high-fat meal, with elevated postprandial lipemia, a strong upregulation of GDF-15 expression in coronary artery SMCs (CASMCs) by triglyceride-rich lipoproteins (TRL) was observed [86]. The group of TRLs is composed of chylomicrons and very low-density lipoproteins (VLDLs). TRLs and their metabolites are involved in the pathogenesis of atherosclerosis by modulating inflammation, oxidative stress, and foam cell formation [87], as well as inducing cell proliferation [88] and monocyte chemoattractant protein-1 (MCP-1) expression in SMCs [89]. Likewise, TRLs and their metabolites have been detected in atherosclerotic plaques [90].
Hence, more research projects are necessary to understand the direct effects of GDF-15 on VSMCs in the context of atherosclerotic plaque development, progression, and stability.
Myocardial infarction, a condition associated with CAD, is associated with many deaths [91] and shows upregulation of GDF-15 after acute myocardial infarction [92]. GDF-15 is not constitutively expressed in adult myocardium. Cardiomyocytes produce and secrete GDF-15 only in response to oxidative stress, angiotensin II or inflammatory cytokines, ischemia, and mechanical stretch [28]. Increased plasma levels of GDF-15 can be detected in patients suffering from myocardial infarction, or as a result of injury and heart failure [93, 94]. Among other findings, data from the Women’s Health Study show that serum GDF-15 levels are an independent risk indicator for adverse CV events [93]. In this regard, GDF-15 has cardioprotective effects on cardiomyocytes in ischemic tissue and controls the conversion of cardiac fibroblasts to myofibroblasts during the development of fibrosis [92, 95]. Thus, understanding cellular GDF-15 signaling and crosstalk in cardiac metabolism is a research concern.
In vitro experiments with immune cells, ECs, and cardiomyocytes from the ventricle suggest that GDF-15 may act as a survival factor on one side and as an inducer of cell death factors on the other [49, 55, 95, 96, 97, 98, 99, 100]. Using GDF-15 gene-targeted mice, endogenous GDF-15 was shown to protect the heart from ischemic/reperfused (I/R) injury [92]. Similarly, cell culture experiments with recombinant GDF-15 showed that cardiomyocytes are protected from hypoxia-induced ischemic injury via PI3K and AKT-dependent signaling pathways [92]. GDF-15 promotes rapid activation by transient Ser473 phosphorylation of AKT in cardiomyocytes, which is accompanied by an increase Ser136 phosphorylation (inactivation) of the AKT downstream target Bcl-2 antagonist of cell death (Bad) [92] (Fig. 4; Table 4, Ref. [92, 95, 99, 101]), a pro-apoptotic protein of the Bcl-2 family [102]. In addition, other PI3K/AKT-independent pathways may be involved in the autocrine/paracrine effects of GDF-15 [95, 99], with GDF-15 transiently activating ERK1/2 in cardiomyocytes [92, 99], but not p38 or JNK [92] (Table 4).
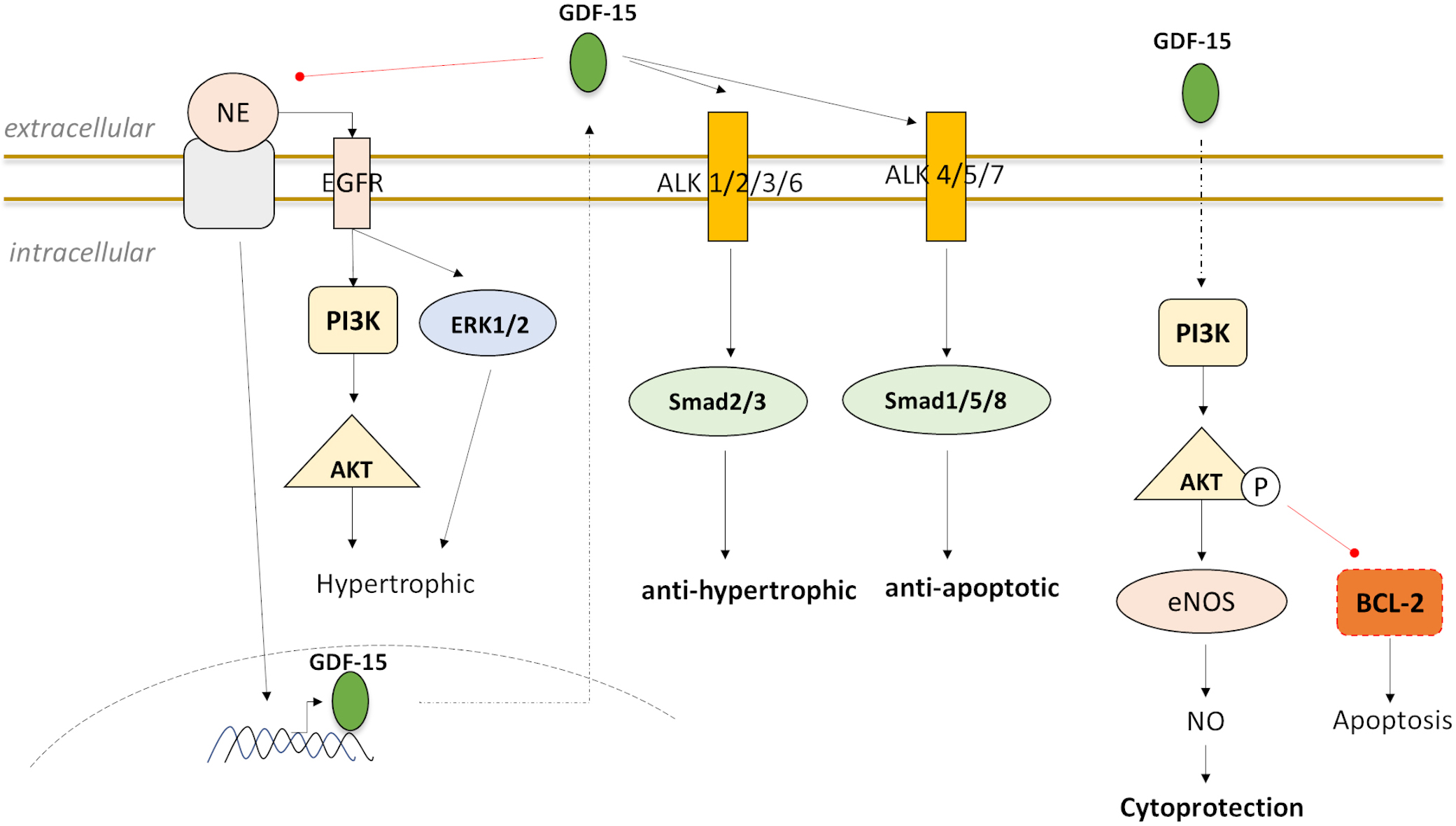 Fig. 4.
Fig. 4.Downstream targeting and signaling of GDF-15 in experimental stress-induced models that stimulate GDF-15 expression in cardiomyocytes and reveal GDF-15 as a cardioprotective via PI3K-AKT, ERK, and SMAD proteins.
| Cell Type | Experimental models | process | Effects and mechanisms | References |
| Ventricular cardiomyocytes from rats | Ischemic injury | Cytoprotective, apoptosis |
AKT, PI3K | [92] |
| Neonatal Ventricular cardiomyocytes from rats | Cardiomyopathy | Cytoprotective, hypertrophic |
R-SMAD2, ERK1/2, AKT | [95] |
| ventricular cardiomyocytes of rat | Heart failure, cardiac remodeling | apoptosis |
PI3K, ERK, and R-SMAD1 | [99] |
| Neonatal rat cardiomyocytes (NRCMs) | Cardiac remodeling | hypertrophic |
EGFR, AKT, ERK | [101] |
Pathological myocardial hypertrophy leads to increased oxygen demand and
decreased contractility of the affected ventricle [103]. This usually results in
heart failure, as well as an increased risk of myocardial infarction [104, 105].
The hypertrophic signaling effect mediated by GDF-15 via the epidermal growth
factor receptor (EGFR), PI3K, AKT, ERK, as well as SMAD proteins is controversial
in this regard [95, 99, 101] (Fig. 4; Table 4). Analyses have shown that GDF-15
attenuates norepinephrine (NE)-induced myocardial hypertrophy as well as
hypertrophy in cultured rat neonatal ventricular cardiomyocytes through induction
of small body size (SMA) and SMAD2/3 phosphorylation and detectable induction of
SMAD1/5/8 phosphorylation [95, 101] (Fig. 4; Table 4). NE is known to induce
oxidative stress resulting in hypertrophy, apoptosis, and intracellular Ca
Recently, 5 years ago (in 2017), four research groups simultaneously identified
the GDF-15 receptor [109, 110, 111, 112]: By using screening arrays of GDF-15 against glial
cell line-derived neurotrophic factor (GDNF) receptors and the orphan GDNF
receptors GFRAL (GDNF receptor
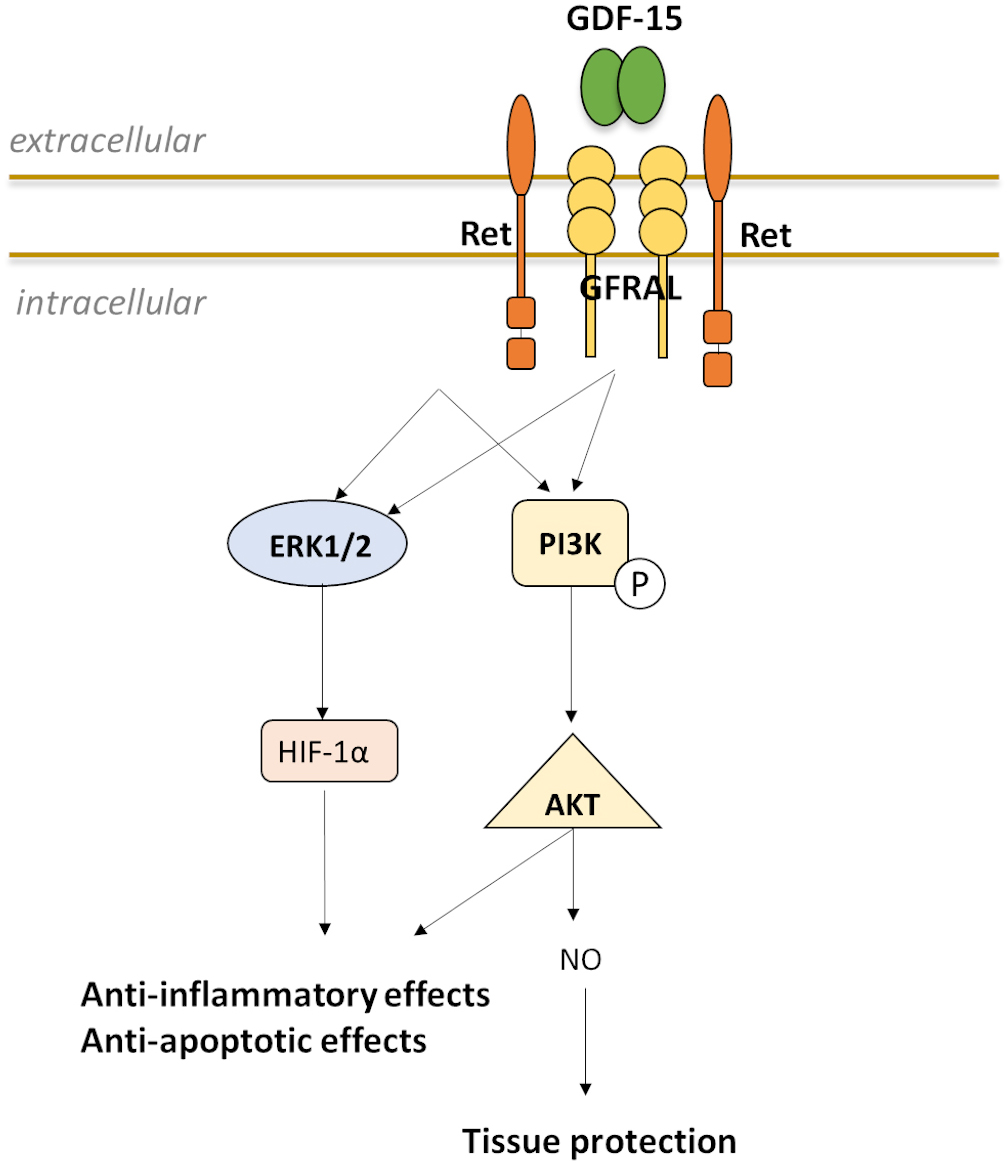 Fig. 5.
Fig. 5.Possible model for induction and interaction of GDF-15 with GFRAL and the coreceptor RET to enable downstream signal transduction.
The importance and relevance of the GDF-15/TGF-ßRII and the GDF-15/NF-kB
pathways in the cardiovascular system are well known [115]. de Jager et
al. [75] examined the signal transduction cascades for GDF-15 and showed that
blockade of TGF-ßRII leads to an abrogation of MCP-1/chemokine (C-C motif) ligand 2 (CCL2) monocyte migration
triggered by GDF-15 [75]. This suggests a crucial involvement of GDF-15 in the
mechanism of atherosclerosis development and progression. As previously
described, the expression of GDF-15 is also upregulated by several
pro-inflammatory stimuli in macrophages, including IL-1ß, IL-2, and
TNF-
Recent studies also reveal an essential role of GDF-15 in the mTOR/autophagy pathway in relation to atherosclerotic progression. GDF-15, in combination with oxLDL, impairs autophagic processes with effect on lipid homeostasis in human macrophages [71]. GDF-15 also appears to be an important factor in regulating autophagy in ECs of atherosclerotic lesions [48], with impaired endothelial autophagy in hypercholesterolemic mice abrogating the antiatherogenic effect of blood flow-induced-shear stress, thereby exacerbating the burden of atherogenic plaques and enhancing inflammatory responses [117].
These signaling pathways provide evidence that targeting the pathophysiological activity of GDF-15 may provide novel therapeutic agents for CAD patients. Thus, targeting the GDF-15 pathway is the focus of new therapeutic approaches to combat CAD.
Early identification of high-risk individuals with CVD is of great importance and could allow timely decisions on preventive measures. Conventional risk factors are enabled for only about half of CAD prevalence. Therefore, it is essential to search for new measurable humoral and genetic markers to improve cardiovascular risk assessment and therapeutic interventions in CAD. GDF-15 is considered one of the most recent promising humoral biomarkers of cardiovascular risk in clinical practice.
Based on the review, from a scientific standpoint, the research perspective is to discover the receptor(s) in the CV system and downstream signaling pathways as the top priority to decipher the activity of GDF-15 in a cell-specific manner. Depending on cell state, cell type, and microenvironment, GDF-15 appears to have both, beneficial and detrimental effects. Clinical studies suggest that patients with elevated GDF-15 levels may benefit from anti-inflammatory, anti-oxidant, or anti-aging therapies. In some studies, increased plasma GDF-15 concentrations over time have already provided strong evidence for poorer prognosis in patients with CAD or heart failure. To date, the reference concentration of GDF-15 in plasma for the healthy general population is not entirely well defined. In this context, further assessment of the effects of environmental and lifestyle factors on GDF-15 concentrations over the life course would provide important insights. Finally, targeted interventions that reduce GDF-15 concentrations could be associated with better health.
ACS, acute coronary syndrome; ALAT, alanine aminotransferase; ASAT, aspartate
aminotransferase; BAD, Bcl-2 antagonist of cell death; BMP, bone morphogenetic
protein; CABG, coronary artery bypass grafting; CAD, coronary artery disease;
CASMCs, coronary artery SMCs; CB-ECFCs, cord blood colony-forming cells; CCL2, chemokine (C-C motif) ligand 2; CCN2,
connective tissue growth factor 2; CHD, coronary heart disease; CRP, C-reactive
protein; cTnT, cardiac troponin-T; CVD, cardiovascular disease; EC, endothelial
cell; ECFCs, endothelial colony forming cells; EGFR, epidermal growth factor
receptor; EndMT, endothelial-to-mesenchymal transition; ERK, extracellular
signal-regulated kinases; GAS1, growth-arrest-specific 1; GDF-15, growth
differentiation factor-15; GDNF, glial cell line-derived neurotrophic factor;
HAECs, human aortic endothelial cells; HE, Hematoxylin-eosin stain;
HIF-1
Data and materials are available on request.
AS has performed literature research and has drafted and written the manuscript. RK and GB have supported writing and drafting and have critically revised the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Not applicable.
The authors thank Silke Vorwald for the excellent histological images.
Open Access funding provided by the Open Acess Publishing Fund of Philipps-Universität Marburg with support of the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.