
† These authors contributed equally.
Academic Editor: Peter Kokkinos
Although the knowledge of sports cardiology advanced significantly in the recent
years, the molecular mechanisms by which exercise training augments cardiac
performance is poorly understood. Here we aimed at determining left ventricular
(LV) myocardial sarcomeric protein modifications in a rat model of exercise
training and detraining. Young male Wistar rats were divided into exercised (Ex)
and control (Co) groups. Trained rats swam 200 min/day for 12 weeks. Detrained
(DEx) and control (DCo) rats remained sedentary for 8 weeks after completion of
the 12-week-long protocol. Ca
The complex morphological and functional aspects of remodeling evoked by
long-term exercise training is called athlete’s heart [1]. Exercise
training-induced cardiac hypertrophy involves improved systolic and diastolic
ventricular functions [2, 3]. Results of cellular electrophysiological studies
(conducted in isolated cardiomyocytes with intact cell membranes) implicated
characteristic alterations in intracellular Ca
Alterations in myofilament protein phosphorylations (e.g., cTnI, myosin binding
protein-C (cMyBP-C) and titin) have been formerly linked to ventricular systolic
and diastolic dysfunctions in cardiac pathological conditions [6, 7, 8, 9]. Of note, the
troponin protein complex (composed of cTnI, troponin T and troponin C) is central
in the regulation of the cardiac contractile protein machinery, and hence its
molecular alterations may well impact cardiomyocyte force production during
physiological hypertrophy as well [10]. Within the troponin complex cTnI is of
particular interest, as this protein holds several phosphorylation sites (e.g.,
for protein kinase A (PKA) and protein kinase C (PKC)), and phosphorylation of
cTnI can mediate a range of distinct contractile responses [7, 9, 11].
Nevertheless, in athlete’s heart Ca
Here we studied exercise-induced cardiac LV remodeling and its reversion
following detraining in a rodent model that involved long-term swim training
[3, 12]. To limit the number of potential confounding variables, we compared
hypothetical relationships between cardiomyocyte contractile function and
phosphorylations of cTnI, cMyBP-C and titin in animals of identical age-sex
groups with or without exercise [14]. Permeabilized cardiomyocytes aided the
detailed characterization of sarcomere dynamics and site-specific phosphorylation
assays allowed the recognition of changes in the phosphorylation level of cTnI.
In this context, phosphorylation of the PKA-specific Ser-22/23 sites or the
PKC-specific Ser-43 and Thr-143 sites reflect hypothetical alterations in
Young male Wistar rats (Toxi-Coop, Dunakeszi, Hungary) (n = 36, m = 200–225 g)
were housed in standard cages at 22
After acclimation, thirty-six rats were divided into control (Co, n = 9),
exercised groups (Ex, n = 9), detrained control (DCo, n = 9) and detrained
exercised (DEx, n = 9) groups. To induce physiological hypertrophy, Ex and DEx
rats underwent a 12-week-long swim training program and were compared to their
counterparts (Co and DCo rats) as described before [3]. To investigate
reversibility, DEx and DCo rats remained sedentary for 8 weeks after the 12-week
long protocol as documented previously [12]. Rats were euthanized after
completion of the in vivo experiments. Subsequently LV myocardium
samples were collected, snap-frozen and stored at –80
LV tissue samples were mechanically disrupted in isolating solution (ISO, (1 mM
MgCl
Cardiomyocytes were solubilized in sample buffer (containing 8 M urea, 2 M
thiourea, 3% (w/v) sodium dodecyl sulfate (SDS), 75 mM DTT, 50 mM Tris-HCl, pH
6.8, 10% (v/v) glycerol, bromophenol blue, 40
Western immunoblotting was applied to assess site-specific phosphorylation status of cTnI. Separation of cTnI and cMyBP-C was carried out in 4% and 12% polyacrilamide gels, respectively. After PAGE and protein blotting procedure, the membranes were blocked with 2% BSA diluted in PBS containing 0.1% (v/v) Tween 20 (PBST, Sigma-Aldrich, St. Louis, MO, USA) for 30 min, then cTnI phosphorylation-sensitive antibodies were used to determine the levels of PKA- and PKC-dependent cTnI (Ser-23/24 (1:1000), Ser-43 (1:500) and Thr-143 (1:500), Abcam, Cambridge, UK) phosphorylation. The signal was detected with a peroxidase-conjugated anti-rabbit IgG secondary antibody (1:300) (Sigma-Aldrich, St. Louis, MO, USA) in nitrocellulose membranes. Total protein amounts were visualized with super sensitive membrane staining (UD-GenoMed, Debrecen, Hungary). Chemiluminescence (ECL) signals of site-specific phosphorylation of cTnI and cMyBP-C were normalized to a Western immunoblot stain.
Cardiomyocyte force generation was measured with a custom-built system (utilizing the DAQ platform produced by National Instruments, Austin, TX, USA) and recorded by a custom-built LabVIEW (National Instruments) module. Results were evaluated in GraphPad Prism 5.0 (GraphPad Software, San Diego, CA, USA). The number of experiments in each group varied between seven and twelve from three or four different hearts. Western immunoblot assays were performed in triplicates. Intensities of protein bands were quantified by determining the area under the intensity curves by a Gaussian fit using ImageJ (National Institutes of Health, Bethesda, MD, USA) and Magic Plot 3.0.1 (Magicplot Systems, Saint Petersburg, Russia) softwares.
Graphs were created in GraphPad Prism 6.0 software. Differences between groups
were calculated by Student’s t-test. Group descriptions were based on
the mean
Ca
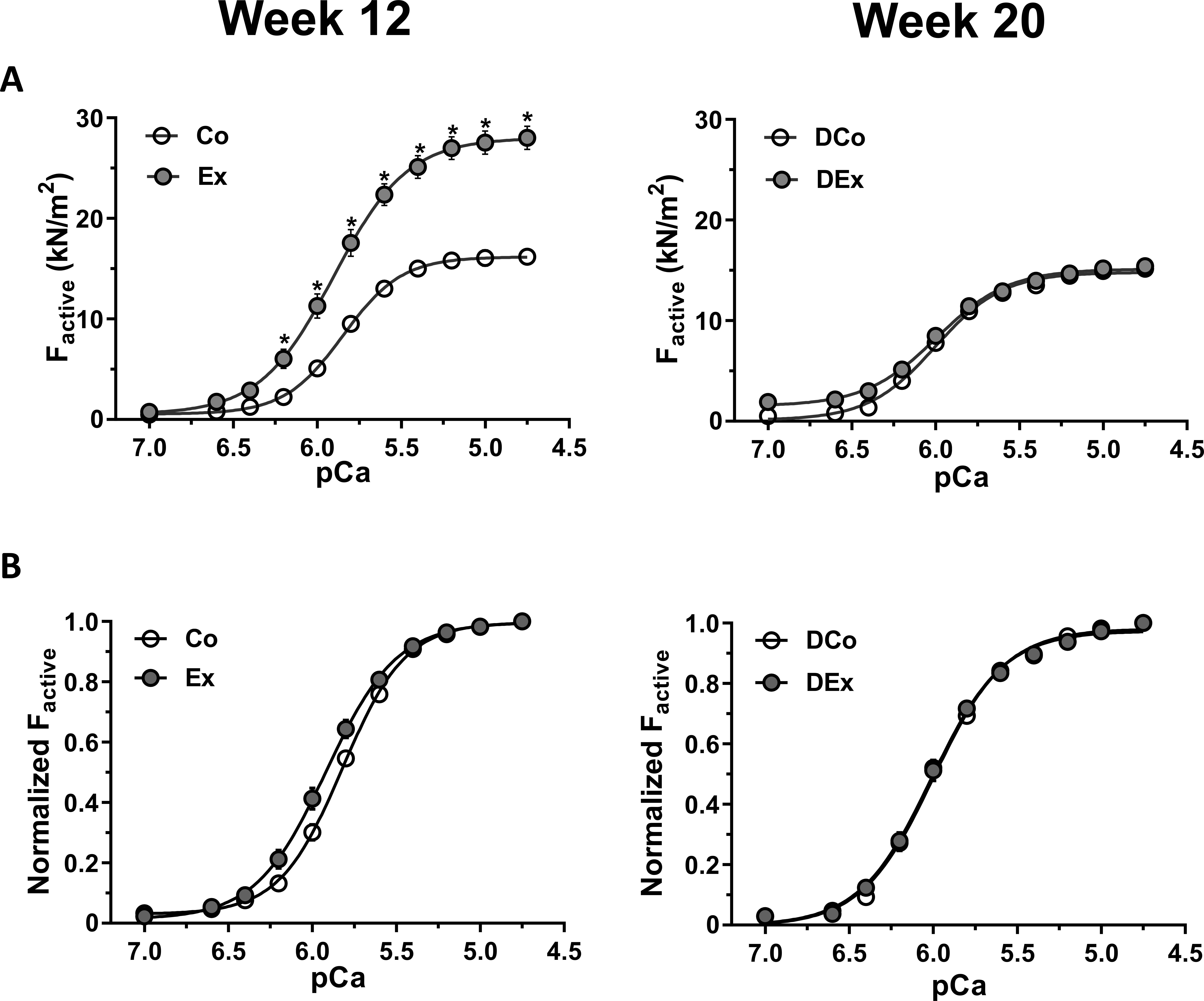 Fig. 1.
Fig. 1.Force production in permeabilized LV cardiomyocyte-sized
preparations following exercise training (week 12) and detraining (week 20). (A)
F
| Week 12 | Week 20 | |||
| Co (n = 11) | Ex (n = 12) | DCo (n = 11) | DEx (n = 9) | |
| F |
15.78 |
28.02 |
15.17 |
15.48 |
| pCa |
5.81 |
5.91 |
5.99 |
6.00 |
| F |
1.49 |
1.75 |
1.70 |
1.50 |
| k |
3.69 |
4.26 |
3.64 |
4.16 |
| cTnI-P (rel.) | 1.00 |
0.66 |
1.00 |
0.91 |
| cMyBP-C-P (rel.) | 1.00 |
1.11 |
1.00 |
0.88 |
| Titin-P (rel.) | 1.00 |
0.91 |
1.02 |
0.99 |
| cTnI |
1.00 |
0.76 |
1.00 |
0.98 |
| cTnI |
1.00 |
0.77 |
1.00 |
1.02 |
| cTnI |
1.00 |
0.99 |
1.00 |
1.03 |
| Values are mean | ||||
Following exercise training, overall phosphorylation level of cTnI decreased
markedly in LV cardiomyocytes. However, this difference disappeared after
detraining. Overall phosphorylation levels of cMyBP-C and titin (not shown) were
similar in permeabilized LV cardiomyocytes in all experimental groups (Fig. 2A,
Table 1). To elucidate the molecular background of increased F
 Fig. 2.
Fig. 2.Phosphorylation levels of sarcomeric proteins of LV
cardiomyocytes following exercise training (week 12) and detraining (week 20).
(A) Overall phosphorylation levels of cardiac troponin I (cTnI) and cardiac
myosin binding protein-C (cMyBP-C) in LV cardiomyocytes in control (Co),
exercised (Ex), detrained control (DCo), and detrained exercised (DEx) rats (from
left to right, representative results). Pro-Q® Diamond staining
was used to detect overall phosphorylation levels of cTnI (upper panel) and
cMyBP-C (lower panel). Total protein amounts were assessed by Coomassie-blue
staining. Phosphorylation levels of myofilament proteins were normalized to
protein amounts and expressed in relative units. (B) Site-specific
phosphorylation levels of cTnI. cTnI phosphorylation levels of the Ser-22/23,
Thr-143 and Ser-43 residues (from top to bottom) were determined by Western
immunoblotting in LV cardiomyocytes. The upper bands reflect the phosphorylation
status of proteins and the lower bands indicate total protein amounts. Bar graphs
illustrate mean
Exercise-induced cardiac hypertrophy, improvements in systolic and diastolic
function and their reversions have been formerly documented for this animal model
by our group [3, 12]. In the present study we characterized myocardial sarcomere
dynamics and sarcomeric protein alterations in LV cardiomyocytes in detail. Major
novelty of the present study is the apparent association between exercise-induced
increases in F
Myocardial contractility is a major determinant of systolic function, that
depends mainly on Ca
The impact of physical deconditioning on exercise-induced cardiac hypertrophy has also been intensively investigated. Although with some uncertainties, most literature data support complete reversibility of exercise-induced alterations after cessation of training, which can be utilized to distinguish it from pathological hypertrophy [12, 15]. Accordingly, the complete reversion of exercise-induced changes in sarcomere dynamics and myofilament protein alterations reflect a physiological type of LV hypertrophy in our model.
Exercise activates the sympathetic nervous system,
Here we also investigated posttranslational modifications at PKC specific cTnI
sites (Ser-43 and Thr-143), that might affect cardiac contractility, F
Taken together, the observed changes in cTnI phosphorylation, F
In contrast to pathological conditions, physiological myocardial hypertrophy is
associated with preserved or even enhanced diastolic function [15]. In this study
we found that F
In summary, this work implicates a close relationship between increased
F
Ca
BB, AO and LM performed the experiments and analyzed the data; AO, TR, BB and ZP conceived and designed the experiments; AT, BM, AO and ZP wrote the paper. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
All procedures were approved by the Ethical Committee of Hungary for Animal Experimentation (permission number: PEI/001/2374-4/2015). All animals received humane care in compliance with the “Principles of Laboratory Animal Care”, formulated by the National Society for Medical Research and the Guide for the Care and Use of Laboratory Animals, prepared by the Institute of Laboratory Animal Resources and published by the National Institutes of Health (NIH Publication No. 86-23, Revised 1996).
Thanks to all the peer reviewers for their opinions and suggestions.
This work was supported by the GINOP-2.3.2-15-2016-00043 project. The project was co-financed by the European Union and the European Regional Development Fund. This work was also supported by project No. TKP2020-IKA-04 and TKP2020-NKA-04 from the National Research, Development and Innovation Fund of Hungary, under the 2020-4.1.1-TKP2020 funding scheme. Project no. K 132623 (to AT) has been implemented with the support provided from the National Research, Development and Innovation Fund of Hungary, financed under the K_19 funding scheme. The research group (AT, ZP) is supported by the Hungarian Academy of Sciences. Project no. NVKP_16-1–2016-0017 (‘National Heart Program’) has been implemented with the support provided from the National Research, Development and Innovation Fund of Hungary, financed under the NVKP_16 funding scheme. The research was financed by the Thematic Excellence Programme (2020-4.1.1.-TKP2020) of the Ministry for Innovation and Technology in Hungary, within the framework of the Therapeutic Development and Bioimaging thematic programmes of the Semmelweis University. This project was supported by grants from the National Research, Development and Innovation Office (NKFIH) of Hungary (K120277 and K135076 to BM). AO was supported by the János Bolyai Research Scholarship of the Hungarian Academy of Sciences.
The authors declare no conflict of interest. Zoltán Papp is serving as one of the Guest editors of this journal. We declare that Zoltán Papp had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Peter Kokkinos.