

1 Division of Cardiology, Department of Medicine, Texas Tech Health Sciences Center, Lubbock, 79430, TX, USA
Abstract
Heart failure (HF) is a complex syndrome that affects approximately 6.5 million adults in the United States. About half of the 6.5 million adults with HF are estimated to be individuals with heart failure with preserved ejection fraction (HFpEF). It is a common cause for poor quality of life, increased health-care resource utilization, and early mortality. HF incidence has risen to epidemic proportions in the recent years. This review attempts to address the epidemiology and pathophysiology of HFpEF. The incidence of HFpEF increased from 48% to 57% from 2000 to 2007 with a slight decrease in 2010 to 52%. The temporal trends in heart failure show an overall stable incidence of HF over the last two decades with increasing incidence of HFpEF and decreasing HFrEF incidence. Many etiologies contribute to the development of HFpEF which makes the treatment very challenging. Pathophysiology of HFpEF is multifaceted stemming from several disease-specific aspects of inflammation and endothelial function, cardiomyocyte hypertrophy and fibrosis, ventricular-vascular uncoupling, pulmonary hypertension and chronotropic incompetence. Hence identifying the risk factors and etiologies is imperative to achieve optimal outcomes in this population. Newer insights into myocardial remodeling have led to an interesting finding of abnormal fibroblasts in HFpEF which are apoptosis resistant and initiate the development of an abnormal myocardial matrix resulting in initiation and progression of the disease. Upregulation of ROS has also been implicated in HFpEF. Further investigation could provide new avenues to target therapeutics specifically to stop initiation and progression of fibrosis.
Keywords
- HFpEF
- diastolic dysfunction
- pathophysiology
- oxidative stress
- myocardial remodeling
Heart failure (HF) is a syndrome which includes multi organ failure. About 6.5 million adults in the United States have heart failure (HF). HF caused 1 in 8 deaths in 2017. Nationally, HF care costs were estimated at $31 billion in 2012 which included costs for health care services, heart failure medications, and missed days of work. Approximately 50% of the 6.5 million adults with HF are estimated to be individuals with heart failure with preserved ejection fraction (HFpEF) as per the Heart and Stroke statistics -2020 update (Virani et al., 2020). HF is a common cause for poor quality of life, increased health-care resource utilization, and early mortality. These sequelae to HF exacerbation episodes are secondary to high frequency of readmissions and repeated hospitalizations. HF has risen to epidemic proportions in the recent years and will steadily increase over the next decades due to aging and longer life spans. As risk factors such as diabetes and obesity increase, HF incidence will also increase thus becoming an eternal burden on the healthcare system.
The diagnosis of HFpEF can be challenging due to its multiple etiologies and the lack of a single diagnostic test. Such challenges in precise diagnosis make estimation of incidence and prevalence more difficult. Currently a wide variety of approaches have been used for diagnosis of HFpEF. Many epidemiological studies use the Diagnosis-Related Code (DRG) and the International Classification of Disease Code [ICD] in addition to ejection fraction (EF) (Dunlay et al., 2017). Other epidemiological criteria proposed are the Framingham, Gothenburg, Boston and the European Society of Cardiology Criteria (Carlson et al., 1985; Eriksson et al., 1987; McKee et al., 1971; Ponikowski et al., 2016). However, these criteria have their own pitfalls as they are based largely on clinical signs and symptoms. It would be worthwhile to combine the criteria to produce one standard set of criteria as they all have overlapping signs and symptoms. Such an approach would produce more uniformity in assessing the epidemiology of HFpEF. More recently natural language processing (NLP) to probe electronic medical records (EMR) is being used. Using artificial intelligence (AI) driven machine-learning algorithms may improve the diagnostic capability of existing systems especially if used to probe EMR in hospitals and cardiovascular imaging centers (Bielinski et al., 2015; Blecker et al., 2016; Shafiq et al., 2017).
Although the overall incidence of HF has reduced approximately 38% in the past
decade (2000 to 2010) the incidence of HFpEF increased from 48% to 57% from
2000 to 2007 with a slight decrease in 2010 to 52%. A recent study analyzed
results from three longitudinal epidemiological cohorts the Framingham Heart
Study (FHS), Prevention of Renal and Vascular End-Stage Disease (PREVEND) study,
and the Cardiovascular Health Study (CHS) (Ho et al., 2016). All of these
studies had cohorts with different baseline ages. Consistent with the existing
literature that the incidence of HFpEF increases with age, the cumulative
incidence of HF (EF
The prevalence of HFpEF has been studied extensively. The two consistent findings have been that the prevalence of HFpEF is higher in women and about 50% of all heart failure patients have preserved ejection fraction (Bursi et al., 2006). From the different studies that currently exist in literature HFpEF prevalence ranges from 31-55% (Bhatia et al., 2006; Brouwers et al., 2013; Ceia et al., 2002; Gerber et al., 2015; Yancy et al., 2005). Such variation has been largely attributed to the differences in EF used to diagnose HFpEF and inherent differences in the study population (van Riet et al., 2016). The prevalence of HFpEF is 2.4 to 3.4 million as estimated in the United States (Vasan et al., 2018).
Risk factors such as coronary artery disease/ischemia, obesity, diabetes,
chronic kidney disease as well as aging contribute to HFpEF. Four clinical
phenotypes such as - aging, obesity, pulmonary hypertension (PH) and coronary
artery disease (CAD) phenotypes have been described based on the risk factors.
This classification describes the heterogeneity in etiology and the need to
target and individualize the treatments to achieve optimum results. Another
classification uses the biological phenogroups. These phenogroups put forth using
machine learning techniques may be better for risk stratification and targeting
therapies. The three phenogroups include the natriuretic peptide deficiency
syndrome group which comprises of younger subjects with moderate diastolic
dysfunction and relatively low to normal levels of the natriuretic peptides; the
extreme cardiometabolic syndrome group consisting of obese diabetics with a high
prevalence of obstructive sleep apnea; and the right
ventricle-cardio-abdomino-renal syndrome group hosting older individuals with
significant chronic kidney disease and cardiopulmonary comorbidities. In terms of
outcomes phenogroups 2 and 3 had the poorest outcomes as compared to phenogroup
1. Cardiovascular outcomes such as cardiovascular death, heart failure
hospitalization and aborted cardiac arrest were worse in phenogroup 3 as compared
to 1. All cause mortality was worse in phenogroups 2 and 3 as compared to
phenogroup 1 (Adamczak et al., 2020; Cohen et al., 2020; Samson et al., 2016; Shah et al., 2015, 2017) The H
The pathophysiology of HFpEF is highly complex due to its multiple etiologies. Initially diastolic dysfunction was the main focus of research. Many other mechanisms have since been identified to play important roles. Some of these include endothelial dysfunction, abnormal ventricular-vascular coupling, abnormal exercise-induced and flow mediated vasodilation, chronotropic incompetence, oxidative stress and PH.
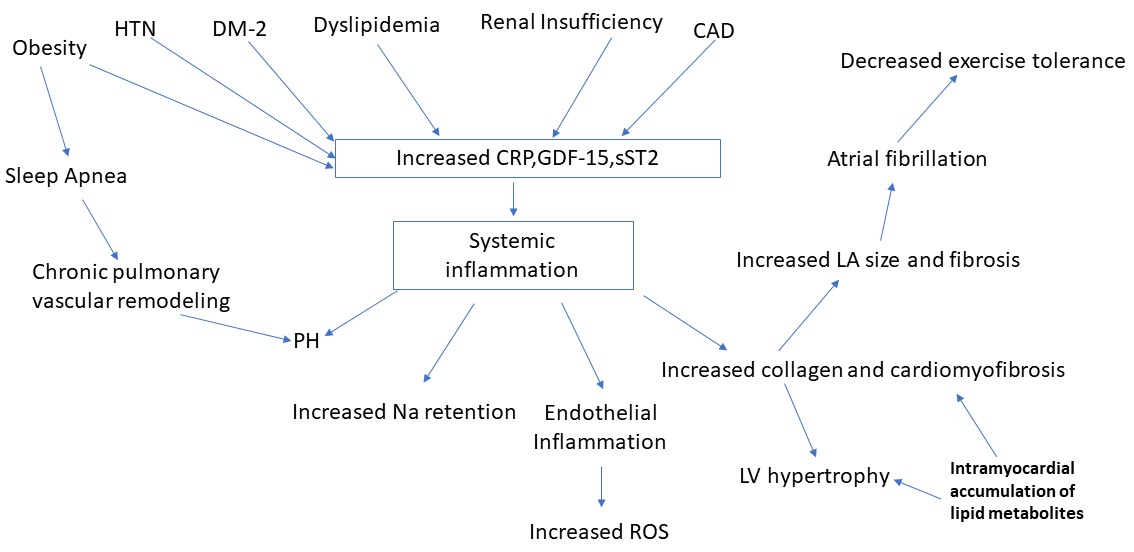
HFpEF is a complex syndrome with multiple etiologies as shown in Fig. 1. Obesity, CAD, hypertension, diabetes, dyslipidemia and chronic renal insufficiency increase systemic inflammation possibly via CRP (C-Reactive protein), GDF-15 (Growth Differentiation Factor-15), sST2 (Soluble suppression of tumorigenesis-2). Systemic inflammation contributes to other pathology such as PH, increased collagen and other matrix protein deposition in turn leading to cardiomyofibrosis and left ventricular hypertrophy (DuBrock et al., 2018; Putko et al., 2014; Zach et al., 2020). Increased fibrosis can lead to atrial and ventricular remodeling to cause arrhythmias such as atrial fibrillation. Such remodeling and pathology eventually leads to decreased exercise tolerance (Fukuta et al., 2019). Obesity is an independent risk factor for sleep apnea which in turn can cause chronic pulmonary vascular remodeling and PH (Ayinapudi et al., 2018; Farr et al., 2016). The cause and effect relationship between several of these factors need to be further investigated due to the complexity of the pathophysiology of HFpEF.
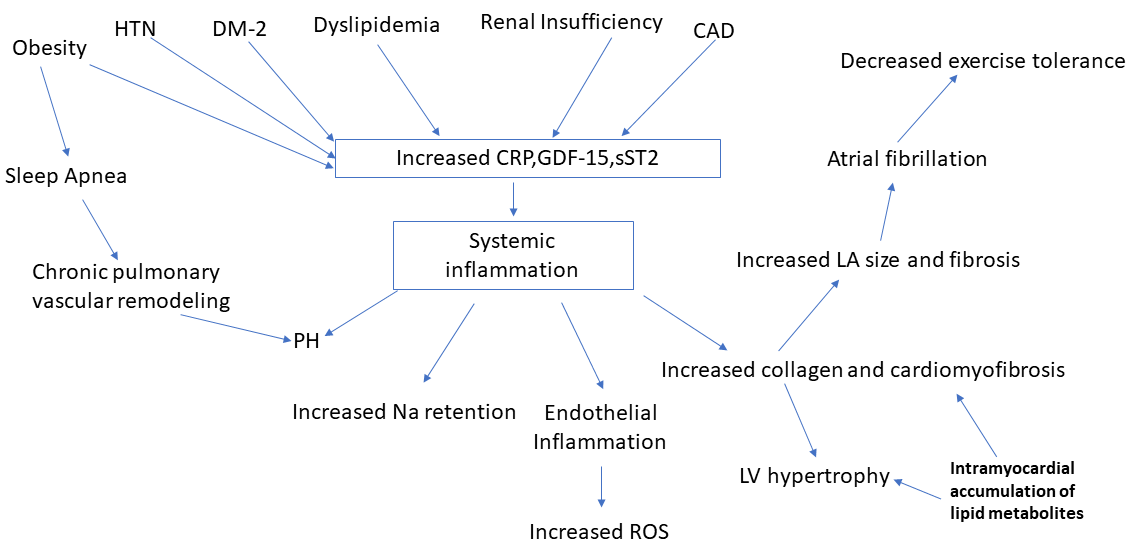 Fig. 1.
Fig. 1.Shows the complex pathophysiology underlying heart failure with preserved ejection fraction. This figure depicts how systemic inflammation is caused by multiple factors which leads to pathology at the molecular and cellular levels. Risk factors such as hypertension, diabetes, dyslipidemia, atherosclerosis and renal insufficiency create a milieu for upregulation of biomarkers for inflammation such as CRP, GDF-15 and sST2 signaling systemic inflammation. Obesity increases systemic inflammation via molecular signaling and also remains one of the most prominent etiological factors for inducing sleep apnea which in turn leads to chronic pulmonary vascular remodeling and PH. PH also results from systemic inflammation making it an important pathology noted in HFpEF. Intramyocardial accumulation of lipid metabolites in diabetes lead to increased collagen deposition and cardiomyofibrosis resulting in LV hypertrophy as well as fibrosis of atrial walls leading to atrial fibrillation and exercise intolerance.
The endothelium has more recently been recognized not just as a barrier between blood and the extravascular tissues but that it is composed of dynamic, highly interactive cells involved in regulating function, physiology and homeostasis of blood vessels. The endothelium prevents platelet and leukocyte adhesion/aggregation, inhibits smooth muscle proliferation, and regulates vascular tone through release of vasoactive substances required for organ perfusion. Nitric oxide (NO) produced from L-arginine by endothelial NO synthase (eNOS) in response to stimuli such as shear stress, cytokines, and platelet-derived factors is an important vasodilatory molecule NO reduces vascular inflammation and atherosclerosis. NO diffuses into platelets and vascular smooth muscle cells and stimulates the soluble guanylate cyclase and activates the cyclic GMP pathway which prevents platelet aggregation and also produces vasodilation. NO diffuses into cardiomyocytes from adjacent coronary microvasculature and modulates cardiac function. Another important function of NO is to mobilize stem cells and progenitor cells responsible for vascular homeostasis and repair. An inverse relationship exists between NO and endothelin (ET). ET is a potent vasoconstrictor and a fine balance of the 2 factors dictate vascular homeostasis (Brutsaert, 2003; Bruyndonckx et al., 2016; Pacher et al., 2007)Cardiovascular risk factors such as smoking, aging, hypercholesterolemia, hypertension, hyperglycemia, and obesity affect the endothelium. Reactive oxygen species (ROS) combine with NO to form peroxynitrite therefore reducing NO availability and tipping the balance to vasoconstriction and a proinflammatory/prothrombotic phenotype. Such a predisposition to vasoconstriction, inflammation and thrombosis disrupts the vascular homeostasis leading to endothelial dysfunction.
Endothelial dysfunction has been implicated in the development of HFpEF
(Paulus and Tschöpe, 2013). The comorbidities observed in HFpEF are
possibly secondary to systemic inflammation resulting in coronary microvascular
endothelial dysfunction and elevated levels of inflammatory cytokines (Bishu et al., 2012; Franssen et al., 2016). Inflammatory cytokines predict the onset of
HFpEF. Inflammation of the coronary microvascular endothelium and reduced
endothelium-dependent vasodilation have been noted in HFpEF. Clinical indices of
endothelial function such as the Flow-mediated dilation (FMD) and Reactive
Hyperemic Index (RHI) are both reduced in HFpEF patients (Borlaug et al., 2010b). Another aspect of importance is that endothelial dysfunction is
directly related to exercise intolerance which is measured by cardiopulmonary
exercise testing and determination of VO
Reduced NO signaling from dysfunctional endothelium influences adjacent cardiomyocytes and cardiac fibroblasts via the sGC-cGMP-PKG pathway resulting in functional and structural cardiac changes such as delayed myocardial relaxation, increased cardiomyocyte stiffness, cardiac hypertrophy, and interstitial fibrosis in patients with HFpEF.
The interaction between HFpEF and endothelial dysfunction results in a vicious cycle further impairing endothelial function. HFpEF causes a systemic inflammatory state with high levels of circulating proinflammatory cytokines and increased production of ROS. This in turn exerts deleterious effects on eNOS expression. Additionally, neurohormonal upregulation in HFpEF leads to increased oxidative stress and upregulation of collagen synthesis. HFpEF worsens systemic endothelial dysfunction leading to progressive heart failure.
Microvascular dysfunction as the cause of HFpEF is a mechanism that can pave the
pathway for therapeutic targets like NO, sarcomeric titin, transforming growth
factor beta (TGF-
Diastolic dysfunction (DD) is a result of myocardial stiffness in the absence of endocardial and pericardial disease. Myocardial stiffness is influenced by the extracellular matrix and the cardiomyocytes. Change in stiffness within the cardiomyocytes is transmitted to the extracellular matrix via matrix proteins. The total amount of collagen type 1 and the extent of collagen cross-linking determines the stiffness of the extracellular matrix. In HFpEF patients, increased deposition of collagen type I results from an imbalance between increased synthesis and decreased degradation (Weber et al., 1993). Collagen synthesis and degradation involve a multistep process in which the procollagen is processed to the collagen type I by proteinases and lysyl oxidase to collagen type I. The degradation is catalyzed by collagenases 9MATRIX metalloproteinases (MMP-1, MMP-8 and MMP-13) and gelatinases (MMP-2 and MMP-9). In HFpEF patients with hypertension or aortic stenosis, a decrease in matrix degradation because of downregulation of matrix metalloproteinases (MMPs) and upregulation of tissue inhibitors of matrix metalloproteinases (TIMPs) has been noted. TIMP-1 appears to be a potential biomarker of HFpEF development in patients with arterial hypertension. Cardiomyocyte stiffness is influenced by titin the cytoskeletal protein (Ahmed et al., 2006; Heymans et al., 2005).
Titin is a giant elastic protein that resides in the cardiomyocytes in two isoforms, N2B (stiffer spring) and N2BA (compliant spring) (Bang et al., 2001) It has been noted in literature that the N2BA : N2B isoform expression ratio is increased in eccentrically remodeled explanted hearts from dilated cardiomyopathy (Makarenko et al., 2004; Nagueh et al., 2004). Such switching of isoforms influences myocardial passive stiffness. Additionally, alterations in the phosphorylation state of titin or formation of disulfide bridges within the titin molecule due to oxidative stress can all induce myocardial stiffness (Borbely et al., 2009; Grutzner et al., 2009; Hidalgo et al., 2009).
HFpEF is characterized by slow LV relaxation reducing LV stroke volume, as heart
rate increases. Cross-bridge detachment and sarcoplasmic reticular Ca
Cardiomyocyte hypertrophy and survival is regulated by matricellular proteins which affects fibroblast function (Schellings et al., 2004). They have been known to improve the quality of the matrix and cardiomyocyte function by binding to collagen, cell surface receptors, and MMPs (Schroen et al., 2004). Their role in the pathophysiology of HFpEF needs further investigation.
PH at rest in HFpEF patients occurs up to 83%. HFpEF patients also show an exaggerated increase in pulmonary artery pressures during exercise. Such an increase in afterload on the right ventricle (RV) in the presence of other risk factors possibly explains the high prevalence of RV dysfunction in HFpEF increasing the morbidity and mortality in this population. Impaired NO-dependent pulmonary vasodilation seen in HFpEF patients is also manifested as reduced exercise-induced pulmonary vasodilation in HFpEF (Andersen et al., 2015; Borlaug et al., 2010a, 2016; Lam et al., 2009; Mohammed et al., 2014).
Pulmonary arterial endothelial dysfunction with higher pulmonary artery pressures were also noted in an animal infarct model of HFpEF in the setting of normal aortic endothelial function and intracardiac pressures (Driss et al., 2000). This suggests that pulmonary vascular endothelial dysfunction precedes systemic endothelial dysfunction in HFpEF. This may be best explained by the fact that the pulmonary circulation is primarily flow-driven versus the pressure-driven systemic circulation and is therefore more susceptible to the shear stress and endothelial dysfunction. In a murine PH model of obese/hypertensive HFpEF rats in which vascular endothelial growth factor receptors were blocked nitrite given orally served as a NO donor preventing the development of PH but unfortunately could not reverse established PH suggesting that long standing PH is fixed and irreversible (Lai et al., 2016; Lam and Brutsaert, 2012).
In a small study of HFpEF patients with PH and severe macrovascular endothelial dysfunction with abnormal FMD an inverse correlation was noted between FMD and pulmonary vascular resistance (PVR) while no correlation was noted when compared with pulmonary capillary wedge pressure (PCWP). This could suggest that longstanding HFpEF is associated with severe endothelial dysfunction in the systemic and pulmonary vasculature (Farrero et al., 2014).
PH can also occur due to reactive pulmonary vasoconstriction and vascular remodeling, which is predominantly mediated by NO, as pulmonary vascular reactivity is maintained by continuous local synthesis of NO. A systemic reduction in NO as noted in HFpEF would lead to pulmonary vascular smooth muscle dysfunction and generate elevated pulmonary pressures and PH (Cooper et al., 1996; Segers et al., 2012).
Pulmonary dysfunction noted in HFpEF patients adds another dimension to the
problem. As pulmonary impairment increases with increase in symptoms it results
in pulmonary edema. However, this finding could be also due to diaphragmatic
dysfunction. Changes in skeletal muscle structure and physiology has been noted
in the diaphragm of HFpEF patients. Fiber Atrophy of myofibers, decreased
oxidative capacity, mitochondrial dysfunction, and increased fati Olson gability
seen in a rat model could explain the effect (Bowen et al., 2015) suggesting
a link between skeletal muscle dysfunction and respiratory abnormalities.
Pulmonary gas exchange is impaired in HFpEF patients, showing O
Vascular remodeling, reactive pulmonary vasoconstriction due to reduced systemic NO bioavailability, impaired diaphragm function, and decreased pulmonary diffusion capacity noted in HFpEF patients contribute to the adverse pathophysiology noted in this population with PH.
Ventricular and vascular stiffening increase with age/hypertension/diabetes and are abnormally elevated in patients with HFpEF (Melenovsky et al., 2007; Paulus and van Ballegoij, 2010) and is strongly associated with impaired exercise capacity (Hundley et al., 2001). Arterial elastance (Ea) and Ees are elevated in HFpEF, resulting in labile blood pressure swings commonly seen in HFpEF, due to exaggerated blood pressure changes seen in preload or afterload changes (Borlaug and Kass, 2008; Kawaguchi et al., 2003). Acute afterload increase in the setting of ventricular-arterial stiffening leads to increases in blood pressure worsening diastolic relaxation and higher filling pressures during stress resulting in exercise intolerance in HFpEF. If ventricular-arterial stiffening is reduced exercise capacity improves in these patients (Borlaug et al., 2007; Chantler et al., 2008; Chen et al., 1999). It should be noted that such ventricular -vascular stiffening increases with age, hypertension and diabetes suggesting that a manifestation of several of the cardiovascular risk factors / comorbidities present in these patients.
Abnormalities in cardiovascular reserve function with exercise stress has been
implicated in HFpEF. due to abnormal venous return, contractility, heart rate,
and peripheral vasodilation. contribute to the pathophysiology. Exaggerated
decrease in chronotropic reserve in HFpEF, is possibly due to deficits in
The beneficial effects of exercise in HFpEF patients is slowly emerging. Current
literature supports significant benefit from training in HFpEF patients
(Edelmann et al., 2011). Exercise training in HFpEF increases VO
Ex-DHF trial secondary analysis showed that inflammatory cytokines (interleukins 1ß, 6, and 10 and tumor necrosis factor alpha) showed no change with exercise but growth hormone releasing peptide ghrelin, which inhibits cardiomyocyte and endothelial cell apoptosis in vitro, increased. Molecular mechanisms underlying exercise induced benefits in HFpEF need further definitive investigation (Conraads et al., 2013; Trippel et al., 2017).
Risk factors for pressure induced hypertrophy (PIH) include hypertension,
advanced age and valvular disease. Longstanding overload initiates mechanical and
neurohormonal upregulation resulting in adaptive changes in the myocardium in the
form of hypertrophy of cardiomyocytes. In the early stages the adaptive response
remains beneficial for maintaining cardiac output but over time it becomes
detrimental leading to hypertrophy of the entire ventricle. At the cellular level
cardiomyocytes increase structural proteins and how abnormal Ca
Early trans differentiation of fibroblast in pressure overload seem to interesting have a strange similarity to fibroblasts associated with cancer. The PIH and cancer associated fibroblasts differ from the reactive fibroblasts noted in physiological healing. These abnormal fibroblasts replace the regular fibroblasts slowly leading to progression of HFpEF with myocardial stiffening. Such abnormal processes also cause solid tumor progression.
Interestingly the PIH fibroblasts and the cancer associated ones share a
molecular signature of expressing
The abnormal fibroblasts cause matrix remodeling by upregulating the matrix metalloproteinases (MMPs). MMP2, MMP9 and MMP14 have been associated with ventricular dysfunction (Chen et al., 2013; Polyakova et al., 2004; Zile et al., 2011). FAP has gelatinase activity and is implicated in degradation of matrix products (Tillmanns et al., 2015). Once the matrix is degraded it is replaced with fibrillary collagens I and III increasing myocardial stiffness (Creemers and Pinto, 2011; Frohlich and Susic, 2012; Spinale, 2007). Domain A fibronectin activates TGF beta resulting in cardiac hypertrophy. Periostin and Tenascin C cause increased fibrosis and progression of HFpEF (Shimojo, 2015; Wu et al., 2016) Impaired diastolic dysfunction in HFpEF can in turn cause activated latent TGF beta expression further increasing cardiac hypertrophy.
The abnormal fibroblasts upregulate the production and secretion of inflammatory
cytokines and growth factors such as IL-1, IL-6, TGF
Cellular and molecular studies have now paved the way to an interesting finding of abnormal fibroblasts in HFpEF which are apoptosis resistant and initiate the development of an abnormal myocardial matrix resulting in initiation and progression of the disease (Oatmen et al., 2019). The fact that it shares molecular markers with cancer associated fibroblasts has opened another therapeutic avenue of using cancer chemotherapeutics to control the fibrosis and its progression (Oatmen et al., 2019).
In 45% of HFpEF patients diabetes exists as a co-morbidity. The characteristics of this population is poorly understood. It is important to understand the pathophysiology when both these conditions exist together to develop personalized medicine. It is interesting that inflammation exists in both conditions but treatments that target inflammation and endothelial dysfunction, such as statins, renin-angiotensin system inhibitors, and phosphodiesterase-5 inhibitors do not sem to be effective suggesting that the pathophysiology that exists in DM is different from those operating in HFpEF (Parikh et al., 2018).
Cardiomyopathy secondary to lipid toxicity is unique to diabetic cardiomyopathy.
Dyslipidemia, increased body mass index and insulin resistance lead to
mitochondrial dysfunction and altered energy metabolism. At the biochemical level
toxic lipid accumulation, membrane lipid remodeling of the cardiac myocytes,
abnormal Ca
Lipid metabolites such as triacylglycerols and ceramides accumulate in the intramyocardial space in obesity and diabetes. This leads to augmented cardiac myocyte apoptosis, fibrosis, impaired contractility and poor diastolic filling. One of the etiologies proposed for intramyocardial accumulation is the downregulation of peroxisome proliferator activated receptor alpha and downregulation of beta oxidation of fatty acids noted in heart failure patients with obesity and diabetes. Further investigation is needed in this area to define clinical relevance for identifying therapeutic targets (Fukushima and Lopaschuk, 2016; Lopaschuk et al., 2010).
Diabetes and HFpEF together cause greater morbidity and mortality. In this population the GWTG-HF registry shows worse in-hospital/post-discharge morbidity, longer length of stay, and higher 30-day all-cause/HF readmissions (McHugh et al., 2019). The pathophysiology of DM in HFpEF patients possibly occurs via increased sodium retention, neurohumoral activation, volume overload, and upregulation of the sodium-glucose cotransporter-2 (SGT2) mechanisms because SGT2 inhibitors decrease volume overload and reduce readmissions (McHugh et al., 2018). DM propagates systemic inflammation in HFpEF patients via multiple pathways such as fatty acid oxidation, decreased nitric oxide availability, and increased AGE. Therapies that target these pathophysiological mechanisms using antihyperglycemic drugs may decrease the progression of remodeling and improve mortality. Graded exercise regimens and targeted therapies may improve skeletal muscle oxygen utilization/exercise tolerance/quality of life (Gandhi et al., 2016). A combination of therapies targeted at different aspects of both the co-morbidities may improve outcomes in this population. Further research is required in this area.
The role of estrogens may be multifold in attenuating progression of HFpEF. It has an important role in reducing oxidative stress/free radical production, endothelial dysfunction, inflammation, regulating the renin-angiotensin aldosterone pathways, and decreasing fibrosis and hypertrophy via upregulation of the atrial and brain natriuretic peptide levels. E2 levels decrease following menopause and is accompanied by changes in body fat, blood pressure, lipid levels influencing cardiovascular risk in postmenopausal women (Matthews et al., 2009). Therefore, strategies targeting E2 could be beneficial in risk factor modification. However, the risks of hormone therapy differ depending on the drug type, dose, duration, administration route, and initiation time, pre-existing cardiac disease and genetic variations. Considering that estrogen has a versatile action on all the cardiovascular risk factors it may have a preventive role in development of HFpEF (Khalil, 2013). Prospective clinical trials and future studies to balance the risk versus benefit to avoid potentially unwanted effects or abnormal hormone-receptor interactions will help. AI using silico models (Cui et al., 2018) also may help in predicting responses. Since HFpEF is more prevalent in women than their male counterparts, role of estrogen in the pathophysiology of HFpEF should be an active area of investigation (Sabbatini and Kararigas, 2020).
HFpEF is increasing in prevalence and relatively high mortality. Many etiologies contribute to the development of HFpEF which makes treatment very challenging. Pathophysiology of HFpEF is multifaceted, stemming from several disease-specific aspects of inflammation and endothelial function, cardiomyocyte hypertrophy and fibrosis. Hence identifying the risk factors and etiologies is imperative to achieve optimal outcomes in this population. Some comorbidities such as sleep disordered breathing is common in HFpEF patients and is associated with worsening diastolic dysfunction. However, a cause and effect relationship and appropriate treatment has not been definitively established (Khattak et al., 2018). As further research into cellular and molecular basis of fibrosis and inflammation became more lucid, targeted therapies for HFpEF would become more apparent. Additionally, role of comorbidities such as diabetes is an important area of research in this population. The role of estrogen in post-menopausal women needs to be better understood to prevent progression of HFpEF in this population.
Nandini Nair was responsible for the conception of ideas presented, writing and the entire preparation of this manuscript.
No funding was obtained for this work.
The author declares no conflicts of interests.