
It is known that functional defects of GATA binding protein 5 (GATA5), an important member of GATA transcription factor family, could cause multiple congenital defects. However, the mechanisms of this transcription factor in cardiovascular diseases are still little known. Finding a genetic approach should help with understanding the possible roles of GATA5 in different cardiovascular diseases and purpose it as a possible therapeutic agent. Hence, this review is divided into three chapters to summarize the roles and main regulatory mechanisms of GATA5 in hypertension, arrhythmia and congenital heart disease, respectively. In each chapter, this review firstly introduces the roles of GATA5 mutations, and then discusses the main regulatory mechanisms of GATA5 in the corresponding diseases (Such as the endothelial dysfunction signaling pathway in the chapter of hypertension, GATA5-NaV1.5 signaling pathway in the chapter of arrhythmia, GATA5-HEY2 and GATA5-Nodal signaling pathway in the chapter of congenital heart disease). Additionally, based on these regulatory networks, it is also speculated that abnormal methylation of the GATA5 gene promoter may lead to cardiovascular diseases such as congenital heart disease. This conjecture is proposed to enrich the regulatory networks of GATA5 and provide a theoretical basis for diagnosis and treatment of cardiovascular diseases.
Rapid economic development has promoted the transformation of the human disease model from communicable disease to non-communicable disease (Du et al., 2019). Cardiovascular disease and cancer are the two most common chronic non-communicable human diseases and are often interdependent (Regulska et al., 2019). Coronary heart disease is one of the major cardiovascular diseases affecting the global population and has been shown to be the leading cause of death in both developed and developing countries (Malakar et al., 2019). In China, the prevalence and mortality of cardiovascular diseases have increased significantly in the past 20 years and are likely to continue to do so into the future (Shen and Ge, 2018). Therefore, it is imperative to further actively explore the mechanism of cardiovascular diseases to stem the ongoing expansion of the cardiovascular diseases burden.
The GATA transcription factor family is divided into two subfamilies GATA1/2/3 and GATA4/5/6. GATA1/2/3 is necessary for the differentiation of mesoderm and ectoderm (including the central nervous and hematopoiesis systems). GATA4/5/6 is involved in the differentiation and development of endoderm and mesoderm, including cardiovascular embryogenesis (Fujiwara, 2017; Lentjes et al., 2016). In vertebrates, each of the six GATA proteins has two zinc finger DNA binding domains, Cys-X2-C-X17-Cys-X2-Cys (ZNI and ZNII), which recognize the sequences (A/T)GATA(A/G) (Lentjes et al., 2016).
GATA binding protein 5 (GATA5) is an important member of the GATA family and regulates many biological processes, such as cell metabolism (Bruun et al., 2014; Zibrova et al., 2017), coronary artery development (Wang et al., 2017; Ying et al., 2017), vascular inflammation, oxidative stress and endothelial function (Messaoudi et al., 2015; Wang et al., 2010) by affecting the expression of bone morphogenic protein (BMP)-4, adenosine 5'-monophosphate (AMP)-activated protein kinase (AMPK), Friend of GATA-2 (FOG-2) and other cytokines. GATA5 deficiency decreases the activity of AMPK and up-regulates the expression of BMP-4, interleukin-6 and intercellular cell adhesion molecule-1 (ICAM-1) (Messaoudi et al., 2015). AMPK promotes the regeneration of coronary artery and exerts a cardioprotective effect under stress conditions such as myocardial ischemia/hypoxia and ischemia/reperfusion (Wang et al., 2017; Zibrova et al., 2017). However, the functional defect of GATA5 decreases AMPK activity (Messaoudi et al., 2015), which weakens the protective effect of AMPK on myocardial cells. This affects the occurrence and development of cardiovascular diseases (Wang et al., 2010). BMP-4 is the earliest marker of atherosclerosis and is considered to be a mechanically sensitive autocrine cytokine that plays an important role in promoting inflammation, atherosclerosis and hypertension (Hong et al., 2016). GATA5 deficiency can lead to hypertension by inducing the production of BMP-4. (Hong et al., 2016; Koga et al., 2013; Messaoudi et al., 2015; Zhang et al., 2014). Therefore, altered GATA5 levels may interfere with the cardiac gene regulatory networks, increasing susceptibility to cardiovascular diseases. Animal experiments and human studies have shown that GATA5 plays an indispensable role in the occurrence and development of cardiovascular diseases such as congenital heart disease, atrial fibrillation and hypertension (Lentjes et al., 2016; Messaoudi et al., 2015; Shi et al., 2014; Wang et al., 2013).
Finding a genetic approach should help with understanding the possible role of GATA5 in different cardiovascular diseases and purpose it as a possible therapeutic agent. Therefore, this review summarizes the roles and main molecular mechanisms of GATA5 in the occurrence and development of hypertension, arrhythmia and congenital heart disease (CHD) (Fig.1). Additionally, based on these regulatory networks, a hypothesis is proposed as a basis for further research.
 Figure 1.
Figure 1.The summary figure of GATA5. GATA5, GATA binding protein 5; CHD, congenital heart disease; SIRT6, Sirtuin 6; Nav1. 5, voltage-gated sodium channel 1. 5; HEY2, Hes-related family basic helix-loop-helix (bHLH) transcription factor with YRPW motif 2; Nodal, Nodal growth differentiation factor; PKA, protein kinase A; AMPK, adenosine 5'-monophosphate (AMP)-activated protein kinase; NF-κB, nuclear factor κB.
Hypertension, with 31 % to 68 % heritability and a global prevalence rate of 25 % to 30 %, is a complex disease determined by both environmental factors and genetic factors (Messaoudi et al., 2015). Hypertension is considered to be a major risk factor for stroke, chronic kidney disease and coronary heart disease, as well as a leading cause of global morbidity and mortality (Guo et al., 2019). Although the findings from the three consortia, Global BP Genetics(GLOBAL BPgen), Cohorts for Heart and Aging Research Genome Epidemiology(CHARGE) and Asian Genetic Epidemiology Network-Blood Pressure (AGEN-BP), represented an important advance in hypertension research, the collective effect of all identified hypertension genes explains only a small fraction (2%) of hypertension heritability (Messaoudi et al., 2015).
The stability of blood pressure is regulated by multiple physiological systems (endocrine, sympathetic, cardiovascular and kidney) (Rafii et al., 2016). GATA5 may balance blood pressure in the kidney and possibly arterial vessels in other organs (Barry et al., 2019). The dysfunction of GATA5 in endothelial cells disrupts this balance by interfering with multiple systems involved in the regulation of blood pressure. Firstly, the absence of GATA5 in endothelial cells promotes renal inflammation and glomerular lesions, which may contribute to the development of hypertension (Messaoudi et al., 2015). Although the mechanism is not clear, it may be related to the secretion of cytokines such as BMP-4 (Tominaga et al., 2011) or the alteration of podocyte mediated by endothelial cells (Daehn et al., 2014; Jefferson and Shankland, 2014). Secondly, in mice, lack of GATA5 can lead to elevated blood pressure, age-dependent terminal organ damage and endothelial dysfunction, which are all characteristics of human hypertension (Messaoudi et al., 2015). Finally, the variations (rs6061245 and rs6587239) of GATA5 sequence are significantly associated with anti-hypertensive medication (Table 1). Rs6587239 is located in the region rich in splicing regulatory sites, which may affect the RNA splicing and the expression of GATA5 (Messaoudi et al., 2015). Therefore, GATA5 could be a susceptible gene for human hypertension.
| References | Mutations | Types of mutations | Cardiovascular phenotypes |
|---|---|---|---|
| Messaoudi et al., 2015 | rs6061245 | Not mentioned | hypertensive |
| rs6587239 | Not mentioned | hypertensive | |
| Wang et al., 2013 | p. W200G | loss-of function | AF |
| Gu et al., 2012 | p. Y138F | loss-of function | AF |
| p. C210G | loss-of function | AF | |
| Yang et al., 2012 | p. G184V | loss-of function | AF |
| p. K218T | loss-of function | AF | |
| p. A266P | loss-of function | AF | |
| Shi et al., 2014 | p. Y16D | loss-of function | BAV |
| p. T252P | loss-of function | BAV | |
| Kassab et al., 2016 | p. T289A | missense variant | AVC malformation |
| p. L233P | missense variant | CoA | |
| p. G166S | missense variant | SV/TOF | |
| p. Y142H | loss-of function | DORV | |
| p. T67P | missense variant | AS, CoA, VSD, PDA, AVC malformation | |
| p. G63A | missense variant | PS | |
| p. R61S | missense variant | VSD | |
| p. S19W | missense variant | ASD, TOF | |
| Wei et al., 2013a | p. R187G | loss-of function | TOF, ASD, PDA |
| p. H207R | loss-of function | TOF | |
| Wei et al., 2013b | p. L199V | loss-of function | VSD |
AF, atrial fibrillation; BAV, bicuspid aortic valve; AVC, atrio-ventricular canal; CoA, coarctation of the aorta; SV, single ventricle; TOF, tetralogy of fallot; DORV, double outlet right ventricle; AS, aortic stenosis; VSD, ventricular septal defect; PDA, persistent ductus arteriosus; PS, pulmonary stenosis; ASD, atrial septal defect; PDA, patent ductus arteriosus
2.1.1 GATA5 deficiency can lead to endothelial dysfunction and hypertension
The latest data show that GATA5, expressed in adult microvascular endothelial cells, is a new blood pressure regulator that regulates many genes and signaling pathways (such as inflammatory pathway) required for endothelial homeostasis and blood pressure regulation (Messaoudi et al., 2015).
Firstly, GATA5 deficiency can lead to oxidative stress and vascular endothelial dysfunction by reducing the activities of protein kinase A (PKA) and AMPK. In terms of mechanism, decreased AMPK activity caused by GATA5 deficiency can increase the activity of 26 S proteasome by changing the phosphorylation status of proteasome subunits and/or interactions between 19 S and 20 S proteasomes. An activated 26 S proteasome exposes the nuclear localization signal of a nuclear factor κB (NF-κB) dimer by degrading the inhibitor of NF-κB α (IκB-α). This allows NF-κB to translocate to the nucleus and induce the transcription of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase subunits (Messaoudi et al., 2015; Wang et al., 2010). Besides, decreased PKA activity caused by GATA5 deficiency can also increase the activity of NADPH oxidase by directly down-regulating the expression of BMP-4 (Hong et al., 2016; Koga et al., 2013; Zhang et al., 2014). These result in oxidative stress and vascular endothelial dysfunction and ultimately promote the occurrence and development of hypertension (Fig.2).
 Figure 2.
Figure 2.Roles of GATA5 in hypertension. GATA5 deficiency can lead to vascular endothelial dysfunction and hypertension by reducing the activities of PKA and AMPK. GATA5, GATA binding protein 5; AMPK, adenosine 5'-monophosphate (AMP)-activated protein kinase; PKA, protein kinase A; NF-κB, nuclear factor κB; IκB-α, inhibitor of NF-κB α; NADPH, nicotinamide adenine dinucleotide phosphate; BMP-4, bone morphogenic protein-4; ROS, reactive oxygen species; IL6, interleukin-6; ICAM-1, intercellular cell adhesion molecule-1; SIRT6, Sirtuin 6; H3K9, histone H3 lysine 9; Nkx3-2, NK3 homeobox 2; PARP-1, poly [ADP ribose] polymerase-1; Bcl-6, B-cell lymphoma-6; VCAM-1, vascular cell adhesion molecule-1; MCP-1, monocyte chemotactic protein-1; MCP-3, monocyte chemotactic protein-3.
Secondly, the functional deficiency of GATA5 up-regulates the expression of pro-inflammatory molecules (such as IL6 and ICAM-1) and decreases vascular nitric oxide (NO) bioavailability, which are expected to contribute to endothelial dysfunction and participate in maintaining the hypertensive phenotype (Messaoudi et al., 2015) (Fig.2).
Finally, poly [ADP ribose] polymerase-1 (PARP-1) inhibits the expression of B-cell lymphoma-6 protein (Bcl-6) by binding to Bcl-6 intron 1. When PARP-1 is phosphorylated by AMPK, Bcl-6 separates from PARP-1 and resumes its expression. Bcl-6 prevents endothelial dysfunction and hypertension by inhibiting the activity of NF-κB and consequently down-regulates the expression of inflammatory genes, including vascular cell adhesion molecule-1, monocyte chemotactic protein (MCP)-1 and MCP-3. However, GATA5 deficiency can inhibit this signal pathway by reducing the activity of AMPK, thus promoting oxidative stress and vascular endothelial dysfunction, eventually leading to hypertension (Gongol et al., 2013) (Fig.2).
2.1.2 Sirtuin 6 (SIRT6) regulates a GATA5-mediated signaling pathway
The epigenetic process mediated by histone deacetylase (HDAC) has attracted much attention in the fields of vascular homeostasis and cardiovascular disease. SIRT6 is a member of the sirtuin family of class III HDACs and is a highly conserved NAD+ dependent deacetylase (Liu et al., 2017; Pillai et al., 2014). Endothelial dysfunction is an important determinant risk factor for the development of hypertension and its complications. SIRT6 is also a key regulator of endothelial function, which inhibits the transcription of the NK3 homeobox 2 (Nkx3-2) by deacetylating histone H3 lysine 9 (H3K9). SIRT6 then induces a GATA5-mediated signaling pathway to prevent endothelial dysfunction and hypertension. Dysfunction of SIRT6 can lead to endothelial dysfunction, hypertension, heart and kidney injury by mediating a GATA5-mediated signaling pathway (Guo et al., 2019) (Fig.2).
Cardiovascular disease is the most common cause of death in the world, with sudden deaths caused by arrhythmias accounting for 50 % of these deaths (Moreau and Chahine, 2018). Atrial fibrillation (AF) is the most common form of arrhythmia observed in clinical practice, as well as being the most common persistent arrhythmia and the main cause of arrhythmia-related hospitalization. It accounts for about 1/3 of hospitalizations due to various arrhythmias. However, AF is a genetically heterogeneous disease with the genetic determinants of most patients still unclear (Gu et al., 2012; Wang et al., 2013). Recently, six significant GATA5 heterozygous mismeaning mutations (p.W200G; p.Y138F; p.C210G; p.G184V, p.K218T and p.A266P) were found in 358 unrelated families with AF (Gu et al., 2012; Wang et al., 2013; Yang et al., 2012) (Table 1), This suggests that the dysfunction of GATA5 may be associated with arrhythmias (including AF).
It has previously been confirmed that mutations of GATA4, GATA6 and NK2 homeobox 5(NKX2-5) have a causal relationship with AF (Yang et al., 2012). GATA4, GATA5 and GATA6 belong to the same GATA subfamily and during cardiac development, especially the process of synergistic regulation of the target gene NKX2-5, their expression and function overlap with each other (Zhang et al., 2007). Thus, GATA5 may promote the formation of a pulmonary myocardial sleeve and shift the pulmonary myocardium to a sinoatrial node-like phenotype by reducing NKX2-5 when dominant negative mutation occurs, hence creating an atrial electrophysiological substrate liable to AF (Yang et al., 2012; Wang et al., 2013).
Additionally, GATA4 and GATA5 bind to the same binding region of the sodium voltage-gated channel alpha subunit 5 (SCN5A) gene promoter and co-activate the transcription of SCN5A gene (Tarradas et al., 2017). Mutations in the SCN5A gene cause a variety of arrhythmias, including Brudaga syndrome, long QT syndrome 3, sick sinus syndrome, cardiac conduction disease, progressive cardiac conduction defect and AF (Han et al., 2018). The SCN5A gene (chromosome 3p21) encodes the alpha subunit of the voltage-gated sodium channel 1.5 (Nav1.5) (Verkerk et al., 2018). Nav1.5 is a giant complex composed of one alpha subunit, four auxiliary beta subunits and many chaperone proteins. It primarily mediates inward sodium current (Savio-Galimberti et al. 2018), which is the main component in the fast depolarization phase after which the excitation-contraction coupling cascade and proper conduction of the electrical impulse is subsequently initiated within the heart (Li et al., 2018). Therefore, GATA5 could regulate cardiac electrophysiological function through a GATA5/SCN5A/NaV1.5 signal pathway and the deficiency of GATA5 function may lead to various arrhythmias (such as AF).
CHD is the most common congenital defect, accounting for 25 % of the total number of structural and functional defects in newborns and 1 % of all newborns (Lage et al., 2012). Severe CHD can result in decreased exercise ability, poor quality of life, pulmonary hypertension, infective endocarditis, thromboembolism, eisenmenger syndrome, heart failure, arrhythmia and even death (Bouma and Mulder, 2017; Brida et al., 2019; El-Assaad et al., 2017; Papamichalis et al., 2019). Although more than 100 gene mutations have been confirmed to be causally related to CHD (Kalisch-Smith et al., 2019), little is known about the specific molecular genetic mechanisms of these CHD susceptible genes.
2.3.1 GATA5 and heart development
Previous studies have confirmed that GATA transcription factors regulate many cardiac genes, such as atrial natriuretic peptide, brain natriuretic peptide, myocyte enhancer factor 2C, NKX2-5, BMP-4, myosin heavy chain 6 cardiac muscle-α and myosin heavy chain 7 cardiac muscle-β (Laforest and Nemer, 2011). GATA5, a member of the GATA family, can coordinate with other factors to regulate the expression of cardiac genes, such as atrial natriuretic factor, connexin 40 and α-actin (Park et al., 2009). GATA5 is a dosage-sensitive regulator of early heart development and acts earlier than GATA4 to regulate the development of heart precursors (Haworth et al., 2008). Moreover, GATA5 also plays an indispensable role in early myocardial differentiation, endocardial cell differentiation, endocardial cushion differentiation and valvular formation (Laforest et al., 2011; Reiter et al., 2001), efficiently directing cardiac fate from embryonic stem cells (Turbendian et al., 2013). In addition, valve development is a complex process, which involves the differentiation and expansion of endocardial cells and their migration after epithelial-to-mesenchymal transformation to form endocardial cushions within the outflow tract and at the atrioventricular canal. GATA5 is an important regulator of valve development, its dysfunction will lead to defective valve morphogenesis and bicuspid aortic valve (Laforest et al., 2011). In zebrafish, GATA5 dysfunction can result in embryonic lethality due to defects in endocardial and myocardial differentiation migration (Heicklen-Klein, et al., 2004).
2.3.2 GATA5 loss-of-function mutations and CHD
Zinc finger transcription factors encoded by the GATA5 gene are necessary for cardiovascular development and structural remodeling. A large number of GATA5 mutations are related to various congenital cardiovascular malformations, including aortic stenosis, pulmonary stenosis, double outlet of right ventricle, tetralogy of Fallot (TOF), bicuspid aortic valve, ventricular septal defect and atrial septal defect (Zhang et al., 2015) (Table 1).
Shi et al sequenced the splice junction sites and coding regions of GATA5 gene in 110 unrelated patients with bicuspid aortic valve and 200 unrelated healthy subjects. Two new heterozygous mutations (p.Y16D and p.T252P) were found in two families with autosomal dominant inheritance of bicuspid aortic valve. Both of them significantly reduced the transcriptional activity of GATA5. This is the first study on the relationship between increased susceptibility to bicuspid aortic valve and GATA5 loss-of-function mutations (Shi et al., 2014). Kassab et al screened the coding exons of GATA5 in 185 patients with different forms of CHD and 150 healthy individuals. Eight missense variants (p.T289A, p.L233P, p.G166S, p.Y142H, p.T67P, p.G63A, p.R61S and p.S19W) were identified. It is confirmed for the first time that human biallelic GATA5 mutations are directly related to the phenotypes of pulmonary stenosis, ventricular septal defect and double outflow right ventricle (Kassab et al., 2016). Wei et al sequenced all the coding regions of GATA5 gene in 130 unrelated TOF patients and 200 unrelated individuals. Two new heterozygous mutations (p.R187G and p.H207R) were found in two families with autosomal dominantly inherited TOF, which significantly decreased the transcriptional activity of GATA5. This is the first report on the link of functionally compromised GATA5 to human TOF (Wei et al., 2013a). Wei et al sequenced the whole coding region of GATA5 gene in 120 unrelated patients with ventricular septal defect and 200 unrelated control individuals. A novel heterozygous mutation (p.L199V) was identified in a patient, which significantly decreased the transcriptional activity of GATA5. this is the first report on the association between ventricular septal defect and GATA5 loss-of-function mutations (Wei et al., 2013b).
2.3.3 GATA5/Connexin 36.7(cx36.7)/NKX2-5/HEY2 Signaling Pathway
As a coactivator of GATA5, NKX2-5 is also involved in the normal cardiovascular development of vertebrates, such as regulation of the number of cardiac precursor cells, development of the conduction system and valve formation (Anderson et al., 2018). GATA4, GATA5 and GATA6 maintain the expression of NKX2-5 by activating cx36.7 (Miyagi et al., 2016), the expression of NKX2-5 can directly up-regulate the expression of the Hes-related family basic helix-loop-helix (bHLH) transcription factor with YRPW motif 2 (HEY2), SCN5A and other genes (Anderson et al., 2018). SCN5A is closely related to arrhythmia (Han et al., 2018). Of note, HEY2 is a member of the bHLH transcription inhibitor family, the non-synonymous mutations of HEY2 are related to cardiac malformations, such as congenital ventricular septal defect (Fardoun et al., 2019). Therefore, GATA5 participates in the regulation of various congenital heart diseases through a GATA5/cx36.7/NKX2-5/HEY2 signaling pathway (Fig. 3).
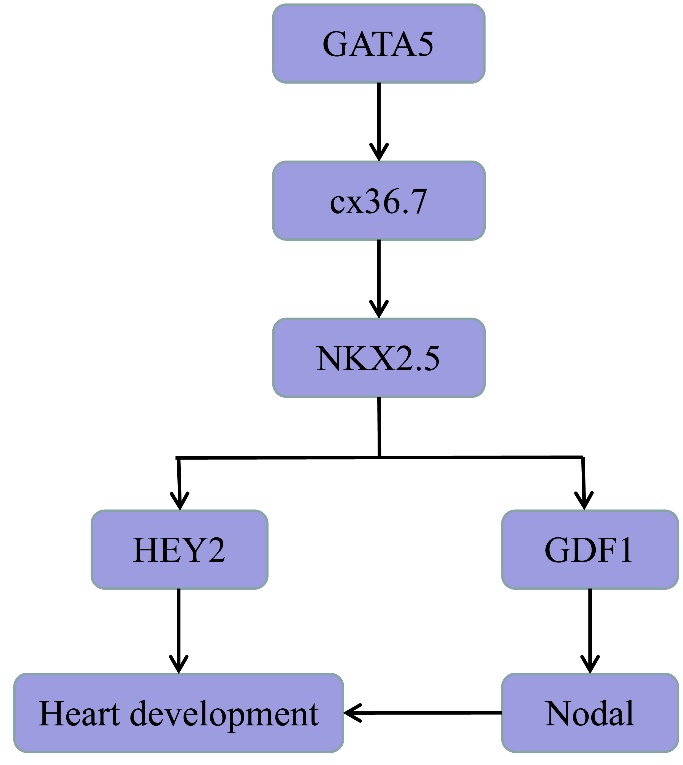 Figure 3.
Figure 3.Roles of GATA5 in congenital heart disease. GATA5 participates in heart development via GATA5/cx36. 7/NKX2-5/HEY2 and GATA5/cx36. 7/NKX2-5/GDF1/Nodal signal pathway. GATA5, GATA binding protein 5; cx36. 7, Connexin 36. 7; NKX2-5, NK2 homeobox 5; HEY2, Hes-related family basic helix-loop-helix (bHLH) transcription factor with YRPW motif 2; GDF1, growth differentiation factor 1; Nodal, Nodal growth differentiation factor.
2.3.4 GATA5/cx36.7/NKX2-5/GDF1/ Nodal Signaling Pathway
As mentioned in section 2.3.3, GATA5 maintains the expression of NKX2-5 by activating cx36.7 (Miyagi et al., 2016). NKX2-5 then binds to the growth differentiation factor 1 (GDF1) promoter and activates its expression in cardiac and non-cardiac cells (Gao et al., 2019). Notably, GDF1 directly interacts with Nodal growth differentiation factor (Nodal) to form a GDF1-Nodal heterodimer, which significantly increases the activity of Nodal (Tanaka et al., 2007). Nodal and its family members are important signal molecules for heart development (Barnes et al., 2016; David and Massagué, 2018; Mohapatra et al., 2009). Therefore, a GATA5/cx36.7/NKX2-5/GDF1/Nodal signal pathway provides another way for GATA5 to participate in the occurrence of various congenital heart diseases (Fig.3).
Functional defects of the GATA5 could lead to the occurrence and development of hypertension, arrhythmia, CHD and many other human diseases through many different signaling pathways. CHD can be caused by environmental factors, oligogenic factors, and/or gene-environment interaction. However, until now, only one-third of CHD cases can be explained by a simple genetic cause. Although compelling evidence that environmental factors can lead to CHD has existed for nearly 80 years, it has not been studied in detail (Kalisch-Smith et al., 2019). It has recently been reported that acute alcohol exposure may ultimately lead to fetal alcohol spectrum disorder (including CHD) by directly affecting the methylation of critical developmental genes (Kalisch-Smith et al., 2019; Ungerer et al., 2013). DNA methylation is one of the most well known epigenetic processes (De Jong et al., 2009). The abnormal methylation of the CpG island in the promoter region of a tumor suppressor gene is the main mechanism of tumor suppressor gene silencing and one of the earliest and most frequent changes in cancer (Belinsky, 2004a, 2005b). GATA5 has been confirmed to be a tumor suppressor gene (Lyon et al., 2007). Abnormal methylation of a GATA5 gene promoter and low level expression of the GATA5 gene have been observed in cancer cells of different types of human cancers, such as lung, pancreatic, esophageal, gastric, colorectal, and retinoblastoma cancers (Akiyama et al., 2003; Guo et al., 2004a, 2006b; Hellebrekers et al., 2009; Livide et al., 2012; Wen et al., 2010).
Therefore, the authors believe that it will be very meaningful to actively explore whether environmental factors lead to the occurrence and development of cardiovascular diseases such as CHD through abnormal methylation of a GATA5 gene promoter. The reasons include: (1) Aberrant methylation of a GATA5 gene promoter can lead to the occurrence and development of various cancers (Akiyama et al., 2003; Guo et al., 2004a, 2006b; Hellebrekers et al., 2009; Livide et al., 2012; Lyon et al., 2007; Wen et al., 2010). (2) Cardiovascular disease and cancer are the two most common chronic human diseases and based on their common etiology and molecular background, they are often interdependent (Regulska et al., 2019). GATA5 mediates the occurrence and development of cardiovascular disease and cancer. For example, the Nodal signaling pathway regulated by GATA5 is not only related to a series of congenital defects, including CHD (Barnes and Black, 2016), but is also involved in maintaining the survival, proliferation, invasion and self-renewal of cancer cell lines (Kalyan et al., 2017; Kirsammer et al., 2014; Wei and Wang, 2018). (3) Epigenetic modifications such as DNA methylation represent the molecular substrate for detrimental environmental stimuli and may lead to cardiovascular disease initiation including CHD (Lorenzen et al., 2012; Vallaster et al., 2012). (4) CHD is primarily caused by non-genetic factors, but the mechanisms associated with environmental factors are still not clear (Kalisch-Smith et al., 2019; Ungerer et al., 2013). This field has great research potential. (5) Gene targeted therapy will inevitably expose the complex in vivo regulatory networks and both its therapeutic effects and side effects are currently unpredictable. But, the interventions of environmental factors are controllable, safe and easy to popularize, to the benefit of many people.
Zhipeng Song wrote the paper. Bo Yan reviewed and edited the manuscript. All of the authors read and approved the final manuscript.
The authors acknowledge funding support from the National Natural Science Foundation of China (81870279). The authors would also like to extend their thanks to the peer reviewers and editors.
The authors declare no conf1licts of interests.