
Cardiovascular disease is still the main cause of morbidity and mortality worldwide. Currently, the frontier of research into cardiovascular disease is the field of non-coding RNA. In this review, information was collected on the use of micro-RNAs as non-invasive biomarkers and their role in pathophysiological processes and therapeutic applications. In the case of microRNA-1 and microRNA-133, the roles and regulatory mechanisms of them are reviewed for arrhythmia, myocardial infarction, diabetic cardiomyopathy, myocardial hypertrophy, cardiomyocyte differentiation, and cell reprogramming. It was observed that microRNA-1 and microRNA-133 do not exist independently, but are two co-transcriptional and cooperative regulatory factors. They have diagnostic value as biomarkers, but also have the potential as therapeutic targets such as for antiarrhythmia and cardiac reprogramming.
Cardiovascular disease and cancer are the two most common chronic diseases in human beings and are often interdependent (Regulska et al., 2019). In association with rapid economic development, disease patterns in China have changed from infectious to non-communicable and cardiovascular diseases have been identified as the main cause of premature (under the age of 70 years) morbidity and mortality (Du et al., 2019). Currently, leading research into cardiovascular-related diseases has shifted from coding genes to non-coding genes with regulatory function. Active exploration of the function of non-coding RNA in the pathophysiology of human diseases will help to enrich understanding of its regulatory networks and provide a theoretical basis for the study of the mechanism of cardiovascular-related diseases (De Windt and Thum, 2015).
In humans, less than 2% of genes can be transcribed into messenger RNA that encodes proteins, while non-coding RNA can be divided into short non-coding RNA (< 200 bp, including microRNA(miRNA) and circular RNA) and long non-coding RNA (lncRNA, > 200 bp) (Shi, 2016). MiRNA (miR)-133 and miR-1, which belong to microRNA, play a significant role in arrhythmia, myocardial infarction, diabetic cardiomyopathy, cardiac hypertrophy, cardiomyocyte differentiation and cell reprogramming.
MiRNA is a kind of endogenous, short (18-24 nucleotides), highly conserved non-coding RNA (Valkov et al., 2019). Primary-precursor miRNAs are transcribed from the genomic region by pol II and subsequently cleaved into precursor miRNAs via Drosha/DGCR8 compound. Precursor miRNAs are then transferred to cytoplasm through exportin-5/Ran-GTP, in which they are further cleaved by the Dicer/TRBP compound and subsequently untwisted into their mature form (Islas and Moreno-Cuevas, 2018). Each miRNA recognizes more than one mRNA, conversely, each mRNA is recognized by multiple miRNAs (Guo et al., 2010). Currently, there are more than 2000 human miRNAs that have been found, which seem to control about 60% of human genes (Shi, 2016). In rare cases, miRs can stimulate the translation of mRNA, but they typically regulate the expression of target genes negatively by promoting mRNA degradation and/or inhibiting mRNA translation (Valkov et al., 2019).
MiRNAs regulate cardiac development, remodeling and regeneration, endothelial function, vasculogenesis and neoangiogenesis through a variety of pathways. They are usually associated with diabetes (miR-126 and miR-320), hypertension (miR-155), dyslipidemia (miR-223), and aging (miR-217 and miR-34a) (Thanikachalam et al., 2018). Many miRNAs, such as miR-17-3p, miR-31 and miR-126 regulate vascular inflammation by regulating the expression of E-Selectin, intercellular cell adhesion molecule-1 and vascular cell adhesion molecule-1 (Staszel et al., 2011). In the process of atherosclerosis, miR-342-5p promotes activation of inflammatory macrophages via protein kinase B pathway (Wei et al., 2013). MiR-124 and miR-135a participate in the regulation of blood pressure by weakening signal transduction by the renin-angiotensin-aldosterone system (Samanta et al., 2016).
In circulation, miRNAs are very stable (28 hours-5 days), easily detected, and mainly transported via carrier molecules such as Exosomes, Argonaute-2 and HDL. Therefore, the circulating concentration of specific miRNA has been used as a biomarker for the diagnosis of a variety of disease conditions including atherosclerosis (miR-130a, miR-21, miR-92, miR-195) (Kwekkeboom et al., 2014; Fung et al., 2019; Wang and Jin, 2018). MiR-499, miR-208, miR-1 and miR-133 are highly expressed in patients with myocardial infarction. Several studies have confirmed the potential of circulating miR-208a and miR-208b, miR-499, miR-133a and miR-1 as sensitive and specific biomarkers of cardiac injury in cardiovascular disease (Islas and Moreno-Cuevas, 2018; Shi, 2016; Fung et al., 2019). Furthermore, circulating miR-133 and miR-1 participate in the mobilization of progenitor cells and other accessory cells from bone marrow to the peripheral circulation, which is the key procedure in the recovery of cardiac function after cardiac ischemic injury (Cheng et al., 2019).
MiR-1 and miR-133 are specifically expressed in heart and skeletal muscle in humans, mice, and zebrafish (Wu et al., 2019). The miR-133 family contains three subfamilies: miR-133a-1, miR-133a-2 and miR-133b. MiR-133a-1 is identical to miR-133a-2 in sequence, whereas miR-133b differs by only 2 nucleotides at the 3' terminus. The miR-1 family also contains three subfamilies: miR-1-2, miR-1-1, and miR-206. MiR-1-1 and miR-1-2 are identical and differ from miR-206 by 4 nucleotides. MiR-133a-1/miR-1-2 and miR-133a-2/miR-1-1 are specifically expressed in heart and skeletal muscle, while miR-206/miR-133b is only expressed in skeletal muscle (Liu et al., 2008). In embryonic stem cells, miR-1 and miR-133 synergistically induce the formation of mesoderm (Xin et al., 2013), whereas their lack of function leads to a variety of heart defects and death (Hagiwara and Kantharidis, 2014). High expression of miR-1 and miR-133 in the injured heart plays a crucial role in cardiovascular pathophysiology (Valkov et al., 2019; Zhang et al., 2019).
Cardiovascular disease is the most common cause of death in the world, with sudden deaths caused by arrhythmias accounting for 50% of these deaths (Moreau and Chahine, 2018). Abnormal upregulation of miR-133 inhibits the expression of the potassium voltage-gated channel subfamily H member 2 (KCNH2), potassium voltage-gated channel subfamily Q member 1 (KCNQ1), and their downstream genes in cardiomyocytes. KCNQ1 is responsible for the function of the slow delayed rectifier K+ current. KCNH2, which encodes the ether-a-go-go related gene(ERG) channel subunit, is responsible for the function of the delayed rectifier K+ current. Inhibition of KCNQ1 and KCNH2 can slow cardiac repolarization, prolong QT interval and eventually induce related arrhythmias (Gui et al., 2018).
Abnormal upregulation of miR-1 suppresses the expression of genes related to the potassium voltage-gated channel subfamily J member 2/Inwardly rectifying K channel 2.1 and the gap junction protein alpha 1/connexin 43 pathway which may then induce arrhythmia by interfering with the K+ current during repolarization (Gui et al., 2017).
Epoxyeicosatrienoic acid (EET) is an endogenous bioactive lipid mediator formed from arachidonic acid through cytochrome P450 epoxygenase enzymes. EETs can be metabolized to dihydroxyeicosatrienoic acids via soluble epoxide hydrolase. Soluble epoxide hydrolase inhibitor (sEHi) increases EET concentration by inhibiting their degradation (Duflot et al., 2014). EETs play an antiarrhythmic role by downregulating abnormally upregulated miR-1 and miR-133 in the myocardium after myocardial infarction (Gui et al., 2018; Liu et al., 2017), but the specific mechanism remains unclear (Fig. 1). Additionally, miR-1 and miR-133 belong to the same gene cluster and can be downregulated simultaneously by sEHis. Therefore, it is speculated that sEHi may downregulate the expression of miR-133 and miR-1 by increasing the inhibition of EETs on the promoters of miR-133 and miR-1.
 Figure 1.
Figure 1.Model of miR-1 and miR-133-mediated regulation of arrhythmia. MiR-1 and miR-133 co-regulate arrhythmia. The red dotted line indicates the regulation that needs to be verified. AA, arachidonic acid; CYP450, Cytochrome P450; EETs, epoxyeicosatrienoic acids; sEH, soluble epoxide hydrolase; sEHi, soluble epoxide hydrolase inhibitor; DHETs, dihydroxyeicosatrienoic acids; miR-133, microRNA-133; KCNQ1, potassium voltage-gated channel subfamily Q member 1; IKs, slow delayed rectifier K+ current; KCNH2, potassium voltage-gated channel subfamily H member 2; ERG, ether-a-go-go related gene; IKr, delayed rectifier K+ current; miR-1, microRNA-1; KCNJ2, potassium voltage-gated channel subfamily J member 2; Kir2.1, inwardly rectifying K channel 2.1; GJA1, gap junction protein alpha 1; Cx43, connexin 43.
The levels of miRNAs (miR-1, miR-133a, amiR-499 and miR-208) in the ischemic myocardium after myocardial infarction are upregulated, but the mechanism is not completely understood. Currently, it is believed that myocardial ischemia can activate the β-adrenoceptor (βAR)/cyclic adenosine monophosphate (cAMP)/protein kinase A (PKA) signal pathway, following activated PKA transfers to the nucleus and phosphorylates cAMP-responsive element binding protein CREB. Activated CREB promotes the transcription of miR-1 and other miRNAs (Shi, 2016; Gui et al., 2017).
After heart injury, miR-1, miR-133a, amiR-499 and miR-208 are rapidly released into the peripheral blood. MiR-499, miR-208, and miR-1 are mainly, and miR133 partially, carried by circulating exosomes. Exosome-miRNAs are selectively and preferentially transferred to bone marrow monocytes where they (miR-1a, not miR-133a, miR-499 or miR-208, directly target the 3' UTR of CXC chemokine receptor 4 (CXCR4)) inhibit the expression of CXCR4 and mediate progenitor cell mobilization. Mobilizing progenitor cells and other accessory cells in bone marrow to peripheral circulation is a key procedure in the recovery of cardiac function after cardiac ischemic injury (Cheng et al., 2019).
Among the four miRNAs upregulated after myocardial infarction, miR133a has the strongest inhibitory effect on the expression of CXCR4, and the concentration of miR133a is the highest in circulation. Although miR133a is only partially carried by exosome, it may have more powerful functions (Cheng et al., 2019). Phosphoprotein 1 secreted by fibroblasts can protect miRNA from degradation in vitro, but it remains to be confirmed whether phosphoprotein 1 plays a role in transporting miRNAs in vivo (Arroyo et al., 2011). Studies have confirmed that circulating high-density lipoprotein and Argonaute2 have the function of transporting miRNAs, but it is not clear whether miR133a is transported in these forms (Tabet et al., 2014; Vickers et al., 2011).
Diabetic cardiomyopathy is featured by contractile dysfunction and cardiac hypertrophy, which ultimately result in heart failure (Hagiwara and Kantharidis, 2014). MiR-133, miR-1, and miR-223 are all highly expressed in diabetic heart and participate in the occurrence and development of diabetic cardiomyopathy (Xiao et al., 2007).
Glucose transporter-4 (GLUT4) is mainly expressed in skeletal muscle and myocardium, as well as brown and white adipocytes, which exhibit an insulin-sensitive glucose transport system (Klip et al, 2019). GLUT4 is reduced in the diabetic heart of patients with type 2 diabetes; however, miR-223 continues to increase in insulin resistant heart tissues and can up-regulate the expression of GLUT4 (Lu et al., 2010). It is suggested that there may be a regulatory loop to restore the expression of GLUT4 in cardiomyocytes exposed to high glucose (Hagiwara and Kantharidis, 2014). On the contrary, miR-133 reduces glucose uptake and metabolism in cardiomyocytes by inhibiting the expression of GLUT4 or Kruppel Like Factor 15 (a transcription factor that drives the transcription of GLUT4) and promoting the occurrence of diabetic cardiomyopathy (Horie et al., 2009).
Cardiomyocyte apoptosis induced by hyperglycemia is another feature of diabetic cardiomyopathy (Yu et al., 2008; Yoon et al., 2005). Mechanistically, both proviral integration site for Moloneymurine leukemia virus-1 (PIM-1) and insulin-like growth factor-1 (IGF-1) can protect cardiomyocytes from mitochondrial dysfunction and apoptosis. However, high glucose can induce the expression of miR-1 and miR-1 directly targets IGF-1 and PIM-1 blocking the cardioprotective effect (Yu et al., 2008; Katare et al., 2011). Additionally, heat-shock protein 60 plays a cardioprotective role by blocking the signal transduction of mitochondrial apoptosis in cardiomyocytes (Shan et al., 2003). High glucose attenuates the cardioprotective effect of HSP60 by increasing the expression of miR-206 and miR-1 in cardiomyocytes, which eventually leads to cardiomyocyte apoptosis and diabetic cardiomyopathy (Shan et al., 2009; Shan et al., 2010).
Myocardial hypertrophy is an adaptive response of cardiomyocytes to various pathological stimuli. It is also one of the main characteristics of myocardial remodeling in the development of heart failure, which itself ultimately leads to irreversible heart damage and increases the morbidity and mortality of cardiovascular diseases. MiR-1 and miR-133 belong to the same gene cluster and play an indispensible role in the regulation of myocardial hypertrophy. They are downregulated in the same way in mice and human myocardial hypertrophy models. As shown in Fig. 2, over-expression of miR-1 or miR-133 in vitro can inhibit myocardial hypertrophy, which suggest that they may co-regulate the expression of cardiac hypertrophy genes (Carè et al., 2007).
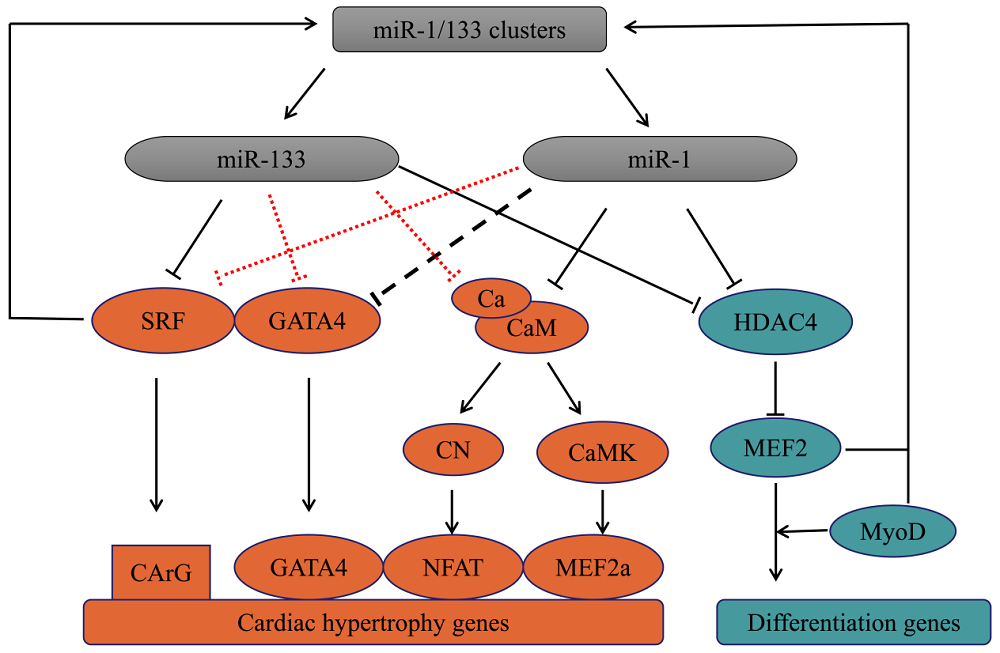 Figure 2.
Figure 2.Model of miR-1 and miR-133-mediated regulation of myocardial hypertrophy and cell differentiation. MiR-1 and miR-133 regulate myocardial hypertrophy and cell differentiation synergistically. The black dotted line indicates indirect regulation, and the red dotted line indicates the regulation that needs to be verified. miR-133, microRNA-133; SRF, serum response factor; CArG, [CC (A/T) 6GG]; GATA4, GATA binding protein 4; miR-1, microRNA-1; Ca, Calcium; CaM, calcium-binding protein calmodulin; CN, calcineurin; NFAT, nuclear factor of the activated T cell; CaMK, calcium-calmodulin dependent protein kinase; MEF2a, myocyte enhancer factor 2a; HDAC4, histone deacetylase 4; MEF2, myocyte enhancer factor 2; MyoD, myogenic determination factor.
Serum response factor (SRF), a widely expressed and highly conserved transcription factor, is a member of the MCM1, Agamous, Deficiens, and SRF-box family. It is directly regulated by miR-133, and also promotes the expression of miR-1 and miR-133 (Chen et al., 2006; Wojciechowska et al., 2017). SRF binds to the promoter of DNA elements containing a CArG box [CC (A/T) 6GG] and promotes the expression of the target genes (Xia et al., 2017). More than 8000 target genes containing CArG sequence have been found and are predicted to dominate the expression of thousands of protein-coding genes (Lighthouse and Small, 2016). More than 200 SRF-dependent genes, which regulate the development, differentiation, proliferation, and apoptosis of cardiomyocytes, have been identified in ventricular cardiomyocytes. (Mikhailov and Torrado, 2016). MiR-133 inhibits myocardial hypertrophy by directly suppressing the expression of SRF (Chen et al., 2006).
Much like SRF, GATA binding protein (GATA)4 is a core transcription factor involved in cardiac development (Schlesinger et al., 2011) and plays an indispensable role in myocardial hypertrophy (Ikeda et al., 2009). GATA4 can directly combine with SRF to promote the expression of hypertrophy genes (Välimäki and Ruskoaho, 2020). GATA4 protein activity is also regulated by post transcription (Välimäki and Ruskoaho, 2020). Additionally, GATA4 also promotes the expression of miR-1 and miR-133 (Schlesinger et al., 2011) and is also regulated by miRNAs in the regulation of myocardial hypertrophy (Ikeda et al., 2009).
Calcium-binding protein calmodulin, another key mediator in hypertrophic signaling pathways, regulates cardiomyocyte hypertrophy through myocyte enhancer factor (MEF)2a and phosphatase calcineurin-nuclear factor of the activated T cell (NFAT) pathways. Like MEF2a and NFAT, GATA4 is also a key regulation factor of cardiomyocyte growth and cooperate to promote the expression of cardiac hypertrophy genes. MiR-1 can inhibit these pathways either directly or indirectly to alleviate cardiomyocyte hypertrophy (Ikeda et al., 2009). As shown in Fig. 2.
Histone acetylation and deacetylation play a key role in regulating gene expression. Histone acetyltransferases catalyze the acetylation of the core histones in nucleosomes, which contribute to chromatin relaxation and transcriptional activation. Histone deacetylase (HDAC) deacetylates the lysine residues of the core histone and promotes chromatin condensation that limits the ability of transcription factors to regulate gene expression. There are four classes of HDACs (I-IV): III enzymes are NAD-dependent enzymes, while I, II and IV are zinc-dependent enzymes. HDAC4 is a class Ⅱ histone deacetylase, which is a target protein inhibited by miR-1 and miR-133a and plays a key role in muscle development, metabolism, differentiation and proliferation (Zhang et al., 2015).
During the metamorphosis of flatfish, the expression of HDAC4 decreased, which reduced its inhibition of MEF2 family transcription factors and promoted cell differentiation (Zhang et al., 2015). During the development of human fetus, the expression levels of miR-133 and miR-1 are directly proportional to the ability of myoblasts to form myotubes (Wu et al., 2019). MiR-133 and miR-1 upregulate the expression of MEF2 by specifically inhibiting HDAC4 and MEF2 then promotes cardiomyocyte differentiation with myogenic determination factor (MyoD). MyoD and MEF2 promote the expression of miR-1 and miR-133 (Chen et al., 2006), and it is speculated that miR-133 and miR-1 synergistically promote cell differentiation (Fig. 2).
During ischemic heart disease, a large number of myocardial cells are lost. Although adult cardiac myocytes slowly self-renew at a measurable rate, it is not sufficient to replace the number of myocardial cells lost after myocardial infarction. Interventional drug procedures and stem cell transplantation improves cardiac function to a certain extent, but the effect is short lived and eventually heart failure develops or even death occurs (Xin et al., 2013). Therefore, cell reprogramming technology based on the regulatory networks of cell proliferation and differentiation began to be explored. Non-myocardial cells in damaged heart can be transformed into functional myocardial-like cells to replace damaged heart tissue and reduce the formation of scars after myocardial infarction, which enhance cardiac function and improve survival rates (Xin et al., 2013).
Cardiac fibroblasts are the main type of cardiac cells, 60%-70% of all cardiac cells are cardiac fibroblasts. They play a supportive and structural role and are considered to be a highly plastic cell type that can be transdifferentiated into functional cardiomyocyte-like cells under certain conditions (Xin et al., 2013; Chiovaro et al., 2015). Snai1 is the main regulator of epithelial-mesenchymal transformation. Overexpression of Snai1 maintains the characteristics of fibroblasts and interrupts cardiac cell reprogramming. MiR-133 induces fibroblasts to transform into functional cardiomyocytes by inhibiting Snai1. The MiR-133/Snai1 pathway is the main pathway for miR-133 participation in cardiac reprogramming (Muraoka et al., 2014). GATA4 is currently the most studied cardiac transcription factor (Rasekhi et al., 2017). Like miR-133, GATA4 also inhibits the Snai1 pathway (Werner et al., 2019), therefore, it is speculated that miR-133 and GATA4 play a synergistic role in cardiac reprogramming. It has been shown that combined injection of miR-133, miR-1, miR-499, and miR-208 into ischemic myocardial tissue induces fibroblasts to reprogram and transform into cardiomyocyte-like cells that then improve cardiac function (Cheng et al., 2019). MiR-1 also induces embryonic stem cells to differentiate into cardiac lineage (Hagiwara and Kantharidis, 2014), but whether miR-1 participate in cardiac reprogramming by inhibiting Snai1 signaling pathway needs to be further verified.
The combination of some transcription factors (such as MEF2C, TBX5 and GATA4; MEF2C, TBX5, Hand2 and GATA4) can also transdifferentiate fibroblasts into functional cardiomyocyte-like cells (Muraoka et al., 2014). Adding miR-133 and miR-1 to the combination of Hand2, MYOCD, TBX5 and GATA4 further improves the myocardial transformation rate of human fibroblasts (Nam et al., 2013). However, it has also been reported that the yield of cardiomyocyte-like cells was higher when TBX5, Hand2, MYOCD and GATA4 were used alone rather than when miRNAs were added. The RNA levels of overexpressed transcription factors TBX5, MEF2C and GATA4 were significantly decreased by the addition of miR-133 and miR-1 (Christoforou et al., 2017). It is speculated that this contradiction may be related to the final concentration of miR-133 and miR-1.
Over the past 50 years, the cardiovascular community has actively explored the impact of environmental factors on cardiovascular disease and reduced the mortality of cardiovascular disease by more than 50%. However since the 1990s, diagnosis and treatment of cardiovascular disease has plateaued and progress has slowed. Despite continuous efforts on environmental factors, the risk of cardiovascular diseases have not further been significantly reduced. The frontier of cardiovascular disease has become the vast field of non-coding DNA (De Windt and Thum, 2015). Both miR-1 and miR-133 in particular, have become recognized as non-coding RNAs with wide function. Previous research exhibited that miR-133 and miR-1 play different roles in cell differentiation and proliferation: miR-133 promotes myoblast proliferation, while miR-1 promotes myoblast differentiation (Chen et al., 2006). But, in reviewing their roles and mechanisms in cardiovascular pathophysiology, we observed that both miR-1 and miR-133 change in the same ways during arrhythmia, myocardial infarction, diabetic cardiomyopathy, myocardial hypertrophy, and cardiomyocyte differentiation. Therefore, it has subsequently been further confirmed that miR-1 and miR-133 do not exist independently, but act as two associated co-transcriptional and cooperative regulatory factors (Wystub et al., 2013; Besser et al., 2014). Furthermore, we also found that miR-1 and miR-133 participate in pathophysiological cardiac processes and play cardioprotective roles through a variety of means. They have a diagnostic value as biomarkers, but also are potential therapeutic targets for antiarrhythmia and cardiac reprogramming.
Following strategies of drug and interventional procedures and stem cell transplantation, cell reprogramming technology has emerged. Compared with stem cell therapy, cell reprogramming bypasses the stem cell-like state and greatly reduces the risk of tumorigenesis (Christoforou et al., 2017). Although it is possible to produce new functional cardiomyocyte-like cells by reprogramming non-cardiomyocytes or inducing the proliferation of existing cardiomyocytes, cell reprogramming is not a mature technology as the efficiency is very low. The phenotype of induced functional cardiomyocyte-like cells is heterogeneous and there is a risk of arrhythmia (Xin et al., 2013). Additionally, the mechanism of miRNAs (including miR-133 and miR-1) in cell reprogramming are still unclear. Therefore, it is particularly necessary to keep on exploring and improving understanding of the signal regulation networks of miRNAs in various pathophysiological processes.
The authors wanted to appreciate the funding from the National Natural Science Foundation of China (81870279). The authors would also like to extend my thanks to all the peer reviewers and editors for their time and consideration.
Bo Yan designed the present study. Zhipeng Song wrote the paper. Rui Gao, Bo Yan reviewed and amended this paper.
The authors declare no conflicts of interests.