
Thanatophoric dysplasia (TD) is a rare form of lethal skeletal dysplasia with underdevelopment of skeleton and dwarfism. The femur is curved in subtype 1, and straight in subtype 2 TD. Other characteristics include a narrow chest, small ribs and hypoplastic lungs. TD is due to activating mutations in fibroblast growth factor receptor 3 (FGFR3), which result in increased receptor activation and alterations in the process of endochondral ossification in all long bones. The aim of the present study was to present the perinatal ultasonographic findings at the 1st and 2nd trimester of a pregnancy and the underlying molecular defect in a fetus with TD type 1, due to a rare mutation in the FGFR3 gene. Ultrasonography performed at the 12w2d of gestation showed increased nuchal translucency (NT). Prenatal karyotype was normal for the XX fetus. Ultrasonography at 17 weeks and 5 days of gestation revealed narrow thorax, abdominal protrusion and a decreased rate of development of the femur (Femur Length, FL < 5th percentile). Molecular genetic analysis to exclude possible overlapping syndromes was performed and revealed de novo c.2419T > G (p. Ter807Gly) (X807G) gene mutation in FGFR3. Fetal autopsy confirmed the prenatal prediction of lethality. We conclude that a fetus with a heterozygous c.2419T > G mutation in FGFR3, presented characteristic biometric parameters and ultrasonographic and autopsy findings consistent with the diagnosis of TD type 1. In addition, the combination of ultrasonography, molecular genetic analysis and autopsy is helpful for the appropriate genetic counselling and perinatal management.
Thanatophoric dysplasia (TD) is the most common lethal form of skeletal dysplasias in fetuses and the affected neonates usually die shortly after birth [1]. The term thanatophoric in Greek means “death bearing” [2]. Maroteaux et al., (1967) described for the first time, thanatophoric dwarfism as a definite discrete entity and differentiated it from achondroplasia [3]. Some years later, thanatophoric dwarfism was renamed TD at the Second International Conference of the Nomenclature of Skeletal Dysplasias [4]. The estimated prevalence of TD in prenatal cases is between 1 : 12,000 and 1 : 20,000 and in births between 1 : 33,000 and 1 : 50,000 [5, 6,7, 8, 9]. TD is classified into two subtypes based on the clinical features of the affected patients: Patients with TD type 1 present with typical curved femurs, short limbs, narrow chest with short thoracic ribs, flattened vertebral bodies and usually absence of a cloverleaf skull. Whereas, patients with TD type 2 present with straight femurs, short limbs, a narrow chest, taller vertebral bodies and a cloverleaf shaped skull. Both types of TDs have additional features including macrocephaly, distinctive facial features, redundant skin fold, brachydactyly and hypotonia [8, 10, 11, 12, 13, 14, 15, 16, 17].
Both types of TDs are inherited autosomal dominant skeletal disorders that are caused by a finite number of mutations in the fibroblast growth factor receptor 3 (FGFR3) gene, which is genomically located on the short arm of chromosome 4 (4p16.3) and consists of 19 exons and 18 introns [8, 9, 13, 18, 19, 20, 21, 22]. Mutations in the FGFR3 gene frequently occur de novo on the paternal allele during male gametogenesis due to DNA copy errors and seems to have a paternal age effect [8, 23]. The FGFR3 protein encoded by this gene is a member of the fibroblast growth factor receptor family. FGFR3 binds fibroblast growth factors and serves as a major negative regulator of linear bone growth [24, 25, 26]. The FGFR3 protein comprises three main parts: an extracellular domain, a transmembrane segment and an intracellular region. The extracellular region of FGFR3 protein includes three immunoglobulin-like (Ig) domains, while its intracellular region is comprised of two tyrosine kinase domains [27, 28]. Ligand binding occurs between IgII and IgIII domains [27, 28, 29, 30].
TD cannot be accurately diagnosed and distinguished from other skeletal dysplasias by prenatal ultrasonography alone. Therefore, molecular genetic analysis of the FGFR3 gene is needed [4, 8, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41]. It has been found that accurate antenatal diagnosis is achieved in only about 30-50% of cases [42, 43, 44, 45]. Therefore, fetal autopsy and postmortem radiography are essential for the confirmation of diagnosis of TDs because they elucidate the exact phenotype, which is not detectable by prenatal ultrasonography [46]. The purpose of the current study was to report the detailed description of the perinatal imaging findings and autopsy characteristics of a fetus with TD type 1 due to a de novo mutation of c.2419T > G (p.Ter807Gly) (X807G) upon DNA analysis.
Α 25-year-old Caucasian gravida 2, para 0, underwent first trimester combined screening (maternal age, nuchal translucency, maternal serum free β-hCG and PAPP-A) at 12w2d of gestation according to the guidelines of Fetal Medicine Foundation. The family history of the couple was unremarkable with no history of skeletal anomalies or other congenital disorders. The mother was healthy and did not suffer from any medical conditions apart from a bicornuate uterus with a past history of one first trimester miscarriage. Her 30-year-old Caucasian husband was healthy and did not suffer from either congenital or acquired conditions. She and her husband did not take any medication or any recreational agents before or during pregnancy. Ultrasound examination was performed using a Voluson machine (General Electric Healthcare System) equipped with a transabdominal 2D 4-8MHz probe. Ultrasound examination showed a single live fetus with fetal crown-rum length (CRL) of 60.3 mm and increased nuchal translucency thickness (NT) of 7.7 mm (Fig. 1). The nasal bone was present. The fetal thorax was narrowed with a prominent abdomen (Fig. 1) but these findings were not evaluated at that time. The amount of the amniotic fluid was normal. The heart rate was 168 beats per minute, a biparietal diameter (BPD) of 19.5 mm, the abdominal circumference (AC) was 57.4 mm and the femur length (FL) 7.5 mm. The estimated risks for trisomy 21, 18 and 13 were 1 : 292, 1 : 57 and 1 : 257, respectively. The patient gave informed consent for amniocentesis at 17w5d of gestation, which was uncomplicated and showed a normal karyotype of a 46, XX fetus. The ultrasound scan revealed narrow, bell-shaped thorax and protrusion of the abdomen (Fig. 2) and shortening of the femur (Fig. 3). The biparietal diameter was 4.04 cm (18w2d) and the head circumference was 14.57 cm (17w2d). The abdominal circumference was 12.36 cm (18w) and the femur length 1.06 cm (13w1d) (< 5th percentile).
 Figure 1.
Figure 1.Prenatal ultrasonographic image at 12w2d of gestation in a fetus with TD type 1 showing increased nuchal translucency.
 Figure 2.
Figure 2.Prenatal ultrasonographic image at 17w5d of gestation in a fetus with TD type 1 demonstrating a narrow chest and protrusion of the abdomen. (A): A narrow chest and protrusion of the abdomen. (B): A protrusion of the abdomen.
 Figure 3.
Figure 3.Prenatal ultrasonographic image of a fetus at 17w5d of gestation with TD type 1 demonstrating short and curved legs with a femoral length measuring 1.06 cm and corresponding to 13w1d of gestation (< 5th percentile).
We therefore suspected a clinical diagnosis of TD, a lethal form of skeletal dysplasia and thus suggested termination of pregnancy. However, the couple was not in contact with the medical staff until 23w2d of gestation. A comprehensive fetal biometric analysis at 23w2d showed: biparietal diameter (BPD) of 58.4 mm and head and abdominal circumferences of 217.2 mm (23w5d) and 178.2 mm (22w5d), respectively. The arms were short with a humeral length of 1.80 cm (corresponding to 16w1d), ulnar length of 1.44 cm (corresponding to 14w5d) and radius length of 1.50 cm (corresponding to 15w) (< 5th percentile) (Fig. 4). The femur length (FL) was 19.6 mm (15w6d) and the tibial length was 1.76 cm (corresponding to 16w) (< 5th percentile) (Fig. 5). The FL/AC and FL/BPD ratios were 0.11 and 0.34, respectively. The average gestational age, including femur length was 21w4d. The femur showed a typical “French telephone receiver” configuration. The nuchal fold was increased (10 mm). The thorax was severely narrowed and irregular with pulmonary hypoplasia. Protrusion of the abdomen was demonstrated (Fig. 6). The amount of amniotic fluid was increased.
 Figure 4.
Figure 4.Prenatal ultrasonographic image of a fetus at 23w2d of gestation illustrating short arms with a humeral length of 1.80 cm (corresponding to 16w1d), ulnar length of 1.44 cm (corresponding to 14w5d) and radius length of 1.50 cm (corresponding to 15 weeks) (< 5th percentile).
 Figure 5.
Figure 5.Prenatal ultrasonographic image of a fetus at 23w2d of gestation illustrating severe shortness of lower limbs with a femur length measuring 1.96 cm (corresponding to 15w6d) and tibial length of 1.76 cm (corresponding to 16 w) (< 5th percentile).
 Figure 6.
Figure 6.Prenatal ultrasonographic image of a fetus at 23w2d of gestation with TD 1 demonstrating a narrow thorax and protrusion of the abdomen.
After extensive genetic counseling, the parents opted for termination of pregnancy by inducing labor, which was accomplished by vaginal administration of misoprostol subsequent to hospital admission and informed consent. She delivered a premature still-born female fetus with severe short-limbed dwarfism. At autopsy, the fetus was weighing 490 g corresponding to the 25th percentile for the 23rd week of gestation (normal weight 600 + 60 g). The crown-rump length was 19.3 cm (normal CRL 20.8 ± 1.9 cm), the head circumferences was 21 cm (normal HC, 15.1 cm), the chest circumference (CC) was 15,8 cm (normal CC, 17 cm) and the circumference of the lower abdomen (AC) was 14.8 cm (normal AC 15.1 cm). The multiple anomalies included macrocephaly, macroglossia, depressed nasal bridge, low-set malformed ears and funnel shaped short neck. Both upper and lower limbs showed rhizomelic and mesomelic brachymelia, humeral and femoral flexion and brachydactyly. The lungs weight was 5.7 g (normal, 14.4 ± 4.3 g) documenting pulmonary hypoplasia. Postmortem radiography revealed macrocephaly with normal ossification, narrow thorax due to short ribs, short bent long bones of the upper and lower limbs, telephone receiver-like short femur, long bone with sharp metaphyseal ends and metaphyseal cupping, iliac bones with horizontal wax and sharp deviations, platyspondyly and hypoplastic vertebral bodies (Fig. 7). Hydrocephalus or clover skull deformity was not noticed. Microscopic examination showed complete disorganization of the epiphyseal growth zone of the cartilage and pulmonary hypoplasia.
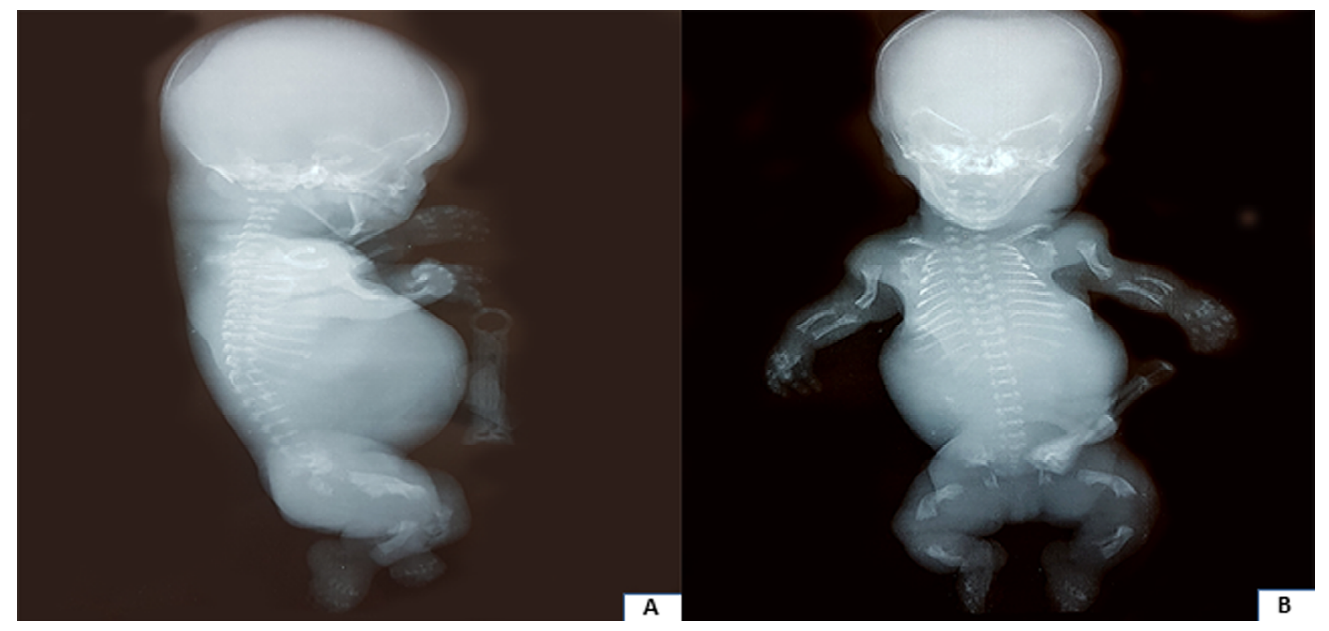 Figure 7.
Figure 7.Postnatal radiography body gram showing narrow chest, short ribs, bell-shaped rib cage, protruding abdomen, short arms, curved femora like "French telephone receiver" and large head. (A) Sagittal view and (B) Coronal view.
Genomic DNA was extracted from fetal tissues and from peripheral venous blood of the parents. Subsequently, conserved coding sequences (CCS) with a size of about 11 Mb, 4,500 of specifically selected genes were enriched with > 150,000 genomic primers designed against the entire human genome (Nextera Rapid Capture Exome, Illumina). Then, sequencing with Illumina NextSeq-500 was performed. The average coverage was 30-50Χ. Comparison was made with the baseline data of HGMD, ClinVar, Exac and HPO. The human reference genome hg19 was used. The molecular genetic analysis showed c.2419 T > G (p.Ter807Gly) heterozygous mutation in the FGFR3 gene. The mutation c.2419T > G (p. 807Gly) in the FGFR3 gene was caused by a T > G transition of nucleotide 2419 on exon 19, leading to a Stop807Gly (TGA > GGA) substitution for proband. This mutation in the chain termination stop codon for FGFR3 gives rise to protein X807G. This fetal genotype was compatible with autosomal dominant disease of lethal type 1 TD. The negative genetic results of the parents demonstrated that the mutation of the fetus was acquired de novo.
The mother gave birth to a healthy female baby twenty one months later weighing 3,060 g at birth.
Skeletal dysplasias constitute a group of heterogeneous disorders affecting growth and morphology of the chondro-osseous tissue [47, 48, 49, 50]. Mutations in FGFR3 are identified in patients with achondroplasia, hypochondroplasia, severe achondroplasia with developmental delay and acanthosis nigricans dysplasia, platyspondylic lethal skeletal dysplasia, San Diego type as well as TD type 1 and type 2 [51]. TD type 1 is caused by FGFR3 missense, insertion or stop codon mutations [13, 18, 19, 22, 52, 53, 54, 55, 56, 57]. Remarkably, these point mutations within the FGFR3 gene which causes TD type 1 constitute gain of function mutations causing ligand-independent overactivation of the FGFR3 receptor by conveying negative signals to chondrocyte and disturbing cartilage function during linear bone growth. Insertion mutations are typically the addition of extra DNA nucleobases to the FGFR3 gene and this results in the creation of unpaired cysteine residues within the extracellular domain of the FGFR3 protein that replaces other amino acids such as Arg, Ser, Gly or Tyr. The newly created cysteine residues allow the formation of disulfide-linked dimers between the extracellular domains of the mutant monomers, hence inducing non-physiological continuous activation of the receptor, which normally requires dimerization for signal transduction. Mutations in the stop codon eliminate the termination codon of the FGFR3 gene [19, 50, 58, 59, 60]. The most frequent mutations of TD type 1 are c.742C > T (p.Arg248Cys), c.746C > G (p.Ser249Cys), c.1108G > T (p.Gly370Cys), c.1111A > T (p.Ser371Cys), c.1118A > G (p.Tyr373Cys) all of which contribute to 90% of the cases [22, 41, 52]. Other mutations of TD type 1 include c.1043C > G (p.Ser348Cys), c.2418G > T (p.Ter807Leu), c.2419T > G (p.Ter807Gly), c.2419T > C (p.Ter807Arg), c.2419T > A (p.Ter807Arg), c.2420G > T (p.Ter807Leu), c.2420G > C (p.Ter807Ser), C2421A > T (p.Ter807Cys), c.2421A > C (p.Ter807Cys) and c.2421A > G (p.Ter807Trp) [34].
TD type 2 is caused almost entirely by a single FGFR3 mutation substituting an A to G at the cDNA nucleotide 1948 (c.1948A > G), causing a lysine to glutamic acid substitution (p.Lys650Glu) in the second portion of the intracellular tyrosine kinase domain of the EGFR3 protein, also resulting in ligand-independent activation [13, 19, 22, 52, 53, 57, 59, 65]. On the other hand, loss of function mutations in FGFR3 cause autosomal recessive camptodactyly, tall stature, scoliosis and hearing loss (CATSHL syndrome) [22]. The FGFR3 signaling pathway requires Snail1 in order to regulate normal chondrocyte proliferation and differentiation and interacts with STAT1 (signal transducers and activators of transcription) pathway, which is involved in the inhibition of chondrocyte proliferation and the MAPK (mitogen-activated protein kinase) pathway, which is involved in chondrocyte differentiation [62, 66].
TD is one type of skeletal dysplasia characterized by severe shortening and deformation of the long bones, macrocrania with frontal bossing, a relatively normal trunk length and thoracic hypoplasia, leading to severe respiratory failure and generally early death [26, 34]. TD type 1 is the most common subtype of TD characterized by a curved femur and occasional cloverleaf skull [51]. TD type 2 is characterized by short but straight femur and cloverleaf skull [51]. Differential diagnosis of TD includes homozygous achondroplasia, achondrogenesis, campomelic dwarfism, rhizomelic chondrodysplasia punctata, severe hypophosphatasia and severe osteogenesis imperfecta [23, 67, 68]. Herein, we presented a case of molecularly proven FGFR3-related TD type 1 initially suspected by abnormal ultrasonographic findings and confirmed with the genetic analysis and autopsy. In our study we pinpointed the specific prenatal ultrasound findings, which are potential markers for the prenatal diagnosis of TD type 1. In the present case, increased nuchal translucency was noticeable at 12w2d of gestation. Increased nuchal translucency was previously reported in TD type 1 [69-72]. In addition, there are reports with prenatal diagnosis of TD at the first trimester of pregnancy [69-74]. In cases with increased nuchal translucency and normal karyotype, the possibility for fetal skeletal dysplasia should be included. In our case, the mother came for amniocentesis at 17w5d of gestation and displayed a normal karyotype of a 46, XX fetus. The narrow fetal thorax and short femur were prominent during the fetal ultrasonographic scans at the second trimester of pregnancy and established the diagnosis of TD. For this reason, we suggested termination of pregnancy. Macrocephaly was not apparent by our ultrasound scans at the second trimester of pregnancy. Also, hydrocephalus or clover skull deformity were not seen. The molecular analysis determined the exact mutation in the FGFR3 gene and the fetal autopsy and the radiological findings confirmed the prenatal prediction of the diagnosis of TD type 1. In a series of 27 cases of lethal skeletal dysplasias by Tretter et al., (1998), 26 were identified antenatally, but only 13 (48%) were given an accurate specific antenatal diagnosis [75]. In addition, in small fetuses with TD type 1 before 20 weeks of gestation, the femoral bowing and the telephone receiver-like configuration may not be prominent [76].
Newborns develop respiratory insufficiency secondary to the narrowed chest cavity [34]. Therefore, almost all neonates with TD require oxygen therapy or mechanical ventilation and most of them die of respiratory failure within one month [77] although 4-9 years’ survivals have been reported [78]. Fetal autopsy and postmortem radiography are essential for the confirmation of TD diagnosis [46]. Postnatal autopsy of the affected fetus revealed disorganized chondrocyte columns, poor cellular proliferation, lateral overgrowth of metaphyses and increased vascularity of cartilage [61, 67]. Terasawa et al., (2019) developed a noninvasive prenatal test using a multiplex PCR system encompassing five mutation hotspots in the FGFR3 gene using cell-free DNA (cfDNA) in the maternal circulation in order to identify the responsible mutations. Paternal samples should be used always as negative control. This system is helpful in early gestation for the distinction between TD and achondroplasia in fetuses presenting with growth retardation and short limbs [79]. Also, this system is helpful in couples with germinal mosaicism for the identification of recurrence of TD [79]. Terasawa et al., (2019) suggested that in cases with positive FGFR3 mutation in maternal cell-free DNA (cfDNA) of peripheral blood plasma, confirmation with chorionic villus sampling or amniocentesis is needed [79].
The prevalence of c.2419T > G among all TD type 1 mutations seems to be 1.6% according to Chen et al., (2017) [41] and 1.7% according to Xue et al., (2014) [22] including platyspondylic lethal skeletal dysplasia, San Diego type (PLSD-SD), since it is due to similar mutations in FGFR3 with TD type 1 [22, 41]. Although the present report is not novel and confirms previously reported findings by Chen et al., (2017) [41], we described one more extremely rare case of TD type 1 in a fetus with a c.2419T > G (p.Ter807Gly) (X807G) mutation in FGFR3 gene and to our knowledge this is the fifth reported case [22, 41]. Comparing our data with those of Chen et al., (2017), our case presented with increased nuchal translucency (7.7 mm) during the first trimester prenatal screening test. Therefore, we suggest that TD type 1 due to a c.2419T > G (p. Ter807Gly) FGFR3 gene mutation should be included in the differential diagnosis in cases with increased nuchal translucency. In addition, comparing our data with those of Chen et al., (2017) we added more information about the ultrasonographic findings at the 17w5d of gestation of this rare case of TD type 1 due to c.2419T > G (p. Ter807Gly) FGFR3 gene mutation.
The nomenclature for the variant c.2419T > G is the rs121913101 (dbSNP, The Single Nucleotide Polymorphism Database) [80, 81]. The mutation c.2419T > G in the FGFR3 gene is caused by a T > G transition of nucleotide 2419 on exon 19 (41). This mutation in the chain termination stop codon in FGFR3 gene eliminates the termination codon. The replacement of a stop codon with a glycine codon at amino acid position 807 results in subsequent extension of the protein by 101 amino acids, denoted p.X807GextX101 [81, 82]. Functional studies by Bonaventure et al., (2007) and Gibbs et al., (2007) showed that an equivalent elongated protein by 141 amino acids at the C-terminal domain (p.X807RextX101) due to a stop codon FGFR3 mutation resulted in constitutive activation of the FGFR3 receptor [83, 84]. Particularly, Gibbs et al., (2007) suggested that the additional C-terminal domain in the mutated FGFR3 protein may function by inducing tyrosine phosphorylation of the FGFR3 receptor in the Golgi apparatus, constitutive activation of this tyrosine kinase FGFR3 receptor and subsequent disruption of bone development [84]. Alternatively, Bonaventure et al., (2007) proposed that hyperactivation of FGFR3 elongated protein is the result of protein stabilization due to a different ubiquitination pattern and reduced degradation [83].
In the current paper we presented the fetal ultrasonographic findings and the autopsy characteristics of a rare case of TD type 1 with a stop codon mutation of c.2419T > G (p.Ter807Gly) (X807G) in the FGFR3 gene resulting in elongation of the FGFR3 protein at the C-terminus by 101 amino acids. Our case reveals that fetuses with TD type 1 may present with increased nuchal translucency. Therefore, TD should be included in the differential diagnosis in cases with increased nuchal translucency during the first trimester prenatal screening test, especially in the presence of normal fetal karyotype. Search for FGFR3 mutations in cell-free DNA (cfDNA) in the maternal circulation is useful for the identification of cases, which will need more search with invasive sampling including chorionic villus sampling or amniocentesis for the definitive confirmation of this fetal malformation. In addition, our case shows that a FGFR3 mutation of c.2419T > G (p. Ter807Gly) (X807G) FGFR3 displays typical features of TD type 1. In our case the fetal autopsy and the postmortem radiography confirmed the diagnosis of TD type 1.
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
MV, AP, AX, AA, ES, AK, AK conceived and designed this case report. MV, AF, AX, VKV, FNV, SA, AA, ES, AK, AK, collected the clinical data and wrote the initial draft of the report. AK performed autopsy. AX, AA, ES, performed genetic analysis. All authors have read and approved the final version of the manuscript.
Written informed consent for autopsy was obtained from the parents.
The patient gave written informed consent for publication of the present case report.
The authors declare that they have no competing interests.