
 , Yushi Zhao 1, Yangyan Sun 1, Wanjiao Chen 1
, Yushi Zhao 1, Yangyan Sun 1, Wanjiao Chen 11 School of Basic Medicine, Gannan Medical University, 341000 Ganzhou, Jiangxi, China
Abstract
Alzheimer’s disease (AD) is the most common cause of dementia. The two major hallmarks of this disease are extracellular amyloid plaques and intracellular neurofibrillary tangles in the brain, accompanied by loss of neurons and synapses. The plaques and tangles mainly consist of amyloid-β (Aβ) and tau protein, respectively. Most of the therapeutic strategies for AD to date have focused on Aβ. However, there is still no effective therapy available. In recent years, the clinical therapeutic failure of targeting Aβ pathology has resulted in increased interest towards tau-based therapeutics. In the current review, we focus on the research progress regarding the pathological mechanisms of tau protein in this disease and discuss tau-targeting therapeutic strategies.
Keywords
- tau-targeting
- microtububle-associated protein tau
- neurofibrillary tangle
- Alzheimer's disease
In 2020, Alzheimer’s disease (AD) was the most common cause of age-related
dementia in China, which is one of the most populous countries, with an estimated
9.83 million people affected (almost 0.7% of the Chinese population) [1].
Worldwide, around 57 million people (more than 0.7% of the general population)
have dementia, with probably 60%–70% of cases being caused by AD [2]. AD is
related to the formation and accumulation of two main protein aggregates:
extracellular amyloid-beta (A
Generally, AD is believed to proceed in a two-stage manner [5]. The first stage
has the characteristics of the emergence and seeded propagation of aberrant
A
Tau is a microtubule-associated protein consisting of six isoforms and plays a physiological role in stabilizing the cytoskeleton and regulating axonal transport through interaction with microtubules [8]. In the adult human brain, tau isoforms are derived from the alternative splicing of exons 2, 3, and 10 [9]. These isoforms differ due to the number of 29-residue inserts encoded by exon 2 and exon 3; tau isoforms having 0, 1, or 2 inserts are called 0N, 1N, or 2N tau, respectively. Alternative splicing of exon 10 generates the 3R- and 4R-tau isoforms, which function differently in the polymerization and stabilization of microtubules. In the normal adult human brain, 3R- and 4R-tau have approximately equal concentrations, but commonly there is a relative overexpression of 4R-tau in pathological conditions [10]. In vitro evidence has shown that alternatively spliced segments encoded by exons 2 and 10 promote aggregation whereas, in contrast, exon 3 depresses it and its efficacy depends on the presence or absence of a fourth microtubule binding repeat domain [11].
In the present review, we discuss tau physiology and pathology, as well as the underlying molecular mechanisms that promote tau pathology, which are mainly involved in post-translational modifications, and further explore the therapeutic strategies that have been tested in animal models and clinical trials to date.
Tau is a microtubule-associated protein (MAP). Normally, tau is mainly distributed within neuronal axons. Functionally, through interaction with microtubules within its repeat domain and flanking regions, tau promotes and stabilizes microtubule assembly. Under normal conditions, tau is natively unfolded, highly soluble, and has little tendency for aggregation, although the tau monomer itself undergoes polymerization and becomes insoluble upon abnormal phosphorylation. In particular, tau regulates microtubular dynamic instability that allows reorganization of the cytoskeleton. Tau knockdown inhibits, whereas tau overexpression promotes, neurite formation [12], suggesting that tau plays a role in axonal elongation and maturation.
Tau plays a potential role in neuronal excitotoxicity [13]. In the brain, tau is primarily expressed within neurons and at low levels in glia, and is also detected outside cells. The reduction of tau by the use of antisense oligonucleotides prevents substantial loss of neurons and reverses the seeding and deposition of pathological tau in hTau-P301S mice [14]. Furthermore, endogenous tau reduction protects against behavioral deficits in human Amyloid Precursor Protein (hAPP)-expressing transgenic mice [15]. Tau has a physiological role in synaptic plasticity. Tau-deficient mice show a long-term depression deficit, which is closely associated with a reduction in synaptic efficiency and subsequently neurodegeneration, rather than long-term potentiation in the Cortical Area 1 (CA1) synapse [16] (see review article [17]). Although existing in axons, tau has a microtubule-independent dendritic function, as seen in truncated tau and tau-/- mice; postsynaptic Fyn localization is decreased, leading to a reduction of N-methyl-D-aspartate (NMDA) receptor phosphorylation, destabilization in the interaction of N-methyl-D-aspartate receptor/postsynaptic density protein-95 (NR/PSD-95), and protection from excitotoxicity [18].
One of the unexpected functions of tau is its nuclear localization [19], which was first found in neuroblastoma cells in 1990 [20], and then later in human brain [21]. Tau occurs in several subcellular compartments, including the cytosol (axons and dendrites) and the nucleus of neuronal and non-neuronal cells. Within neurons, nuclear tau exists mostly in a non-phosphorylated state during cellular stress [22]. In in vitro and in vivo studies under physiological and stress conditions, nuclear tau plays a role in maintaining the integrity of genomic DNA, and cytoplasmic and nuclear RNA [23, 24]. Tau localizes to the nucleus in normal cells in phosphorylated and non-phosphorylated forms; it associates with Transcription termination factor 1 interacting protein 5 (TIP5)—the major subunit of the nucleolar remodeling complex and a key player of heterochromatin stability—localizes to the nucleolus, and represses ribosomal DNA (rDNA) [24]. It is thought that aberrant modifications of tau can change its function and increase the vulnerability of the genome, which leads to neurodegeneration [25]. In fact, researchers are still far from understanding the key role of nuclear tau in the nucleus.
Tau may act as a signaling hub. For example, internalized tau monomer can
trigger increased activation of Extracellular regulated protein kinases1/2
(ERK1/2) [26], which is a key regulator of the behavior, life-cycle, and fate of
cells [27], in addition to having kinase activity. Tau deletion affects ERK1/2
activation and neurite growth [28], suggesting an intracellular signaling role
for tau. Tau may also interfere with Src/Fyn signaling (tyrosine kinase family)
at dendrites. Tau interacts with the phosphatase and tensin homologue protein
(PTEN) and modulates insulin signaling. Recent data suggest that loss of tau
function results in an impaired response of the hippocampus to insulin, due to
altered activities of insulin receptor substrate 1 and PTEN [29]. Interacting
with various phosphatases and kinases, tau may serve as a scaffolding protein to
regulate phosphorylation-based signaling pathways [30]. There is a mechanism by
which pathological modifications of tau induce its conformational changes that,
in turn, promote exposure of its extreme N-terminus, leading to activation of the
protein phosphatase 1-glycogen synthase kinase 3
Intracellular tau may be secreted into the extracellular medium. Once in the extracellular space, tau species can be internalized by neighboring neurons and glia via endocytosis, pinocytosis, or phagocytosis [32]. Under pathological conditions, secreted tau may participate in tau seeding and propagation (discussed later). Tau secretion is involved in vesicular and non-vesicular pathways [33]. Evidence has suggested that tau may be secreted by direct translocation across the cell membrane through interaction with components enriched in the inner leaflet of the plasma membrane mediated by sulfated proteoglycans [34]. Growing evidence has addressed the primary biological mechanisms of tau secretion. It seems that cells secrete tau through three coexisting pathways [35]: (1) translocation across the plasma membrane, (2) membranous organelle-based secretion (exosomes), and (3) ectosomal shedding.
In summary, tau has multifaceted functions in addition to the assembly of microtubules. All these functions are potentially involved in the development of tau pathology and tau-related events.
In AD, tau is post-translationally modified and forms paired helical filaments
(PHFs) that deposit in NFTs in neuronal soma. Toxic tau species ultimately kill
neurons. Unlike extracellular accumulated A
The development of tau pathology is a complex, multifactorial process in which there are multiple targets and specific therapeutic interventions are available. Modified tau per se forms the aggregates in neuronal and/or glial inclusions detectable in the autopsied brain. These modifications contribute to the generation of insoluble fibrous deposits that play an essential role in neurodegeneration [37]. Tau aggregates into PHFs; the repeat domain composes the core of the PHFs, with the short C-terminal and long N-terminal domains surrounding, forming the ‘fuzzy coat’ [38]. In AD, normal tau function is lost and tau is subsequently aggregated according to its modifications, ultimately resulting in the development of NFT pathology [39].
Cognitive decline and behavioral impairment are better correlated with
exacerbated tau pathology and subsequent loss of synapses than amyloid plaques
[40]. Tau phosphorylation may also enhance A
Pathological tau in AD is characterized, to a large extent, by a series of extensive PTMs, including but not limited to hyperphosphorylation, acetylation, sumoylation, truncation, ubiquitination, and glycosylation. Of these PTMs, hyperphosphorylation is one of the earliest and most prevalent modifications, being most closely related to the formation of pathological inclusions. Herein, we address the factors contributing to or mitigating tau modifications and focus on the clinical development of tau-targeting therapies.
Tau hyperphosphorylation is one of the earliest and predominant events in AD. At
least 85 putative phosphorylation sites (Ser, Thr, Tyr) have been identified, of
which
The degree of tau phosphorylation depends on the levels of kinase activity and
the balance between kinases and phosphatases activity within neurons. The primary
tau kinases are as follows: GSK3
GSK3, including two isoforms (
CDK5 is a neuron-specific serine/threonine kinase. Due to the restricted
expression of its activators (p35 or p25) in the nervous system, CDK5 plays a key
role in neuronal migration, neuronal differentiation, synaptic development, and
other functions. CDK5 is involved not only in the phosphorylation of tau but also
in regulating A
Two other kinases (p38-MAPK and DYRK1A), capable of phosphorylating tau, deserve to be discussed here. P38-MAPK is activated at AD lesion sites. Neuronal p38ɑ deficiency ameliorates the symptoms and pathology of AD in an htau-transgenic mouse model [60]. MW181, a brain-permeable inhibitor selective for p38, exerts its effect on tau phosphorylation in vitro and in htau mice [61], indicating that p38 clould serve as a therapeutic target for AD. Overproduction of DYRK1A—a novel tau kinase—observed in postmortem AD brains has been linked to an increased level of tau phosphorylation [62, 63]; DYRK1A also protects against neurodegeneration and behavioral deficits [64]. Recently, specific inhibitors of DYRK1A have been identified, demonstrating the therapeutic potential of targeting DYRK1A in animal models [65, 66].
It is well established that protein phosphatases (PP1, PP2A, PP2B, and PP5) have
the ability to dephosphorylate tau [67]. The most important phosphatase, capable
of dephosphorylating tau, is protein phosphatase 2A (PP2A), accounting for more
than 70% of the total activity of phosphatases. Total PP2A activity has been
shown to be significantly decreased in AD brains. Deficits in PP2A activity are
determined by down-regulation of the PP2A catalytic C subunit at the gene, mRNA,
and protein expression levels in AD. Two important mechanisms (methylation and
inhibition of the activity by endogenous inhibitors known as I1 and I2) regulate
the activity of PP2A [68]. Deregulation of methylation in PP2A can result in loss
of PP2A/B
Tau glycosylation and acetylation are important aspects of PTMs. In the human brain, tau is also modified by glycosylation. O-GlcNAcylation is a dynamic modification of nucleocytoplasmic proteins with the monosaccharide GlcNAc. Altered O-GlcNAcylated protein has been reported in postmortem AD brains [71]. The level of O-GlcNAcylated tau has been observed to be markedly decreased in AD brains, probably attributed to a dysfunction of cerebral glucose metabolism [72, 73, 74]. Increasing evidence suggests that the rapid transition from phosphorylation to O-GlcNAcylation of tau plays a key role in the pathogenesis of AD and the lack of O-GlcNAcylation of tau can induce its hyperphosphorylation [75, 76]. An elevated level of O-GlcNAcylated tau with an O-GlcNAcase inhibitor, leading to a decrease in the level of tau phosphorylation, may successfully improve neurodegeneration in AD mice [77, 78, 79, 80]. Acetylation has been found to enhance tau aggregation in vitro and is commonly seen with modifications at residues Lys and Arg. Acetylation of tau inhibits its degradation and promotes tauopathy [81]. Evidence has demonstrated the role of sirtuin 1, a class III protein deacetylase dependent on nicotinamide adenine dinucleotide, in regulating tau acetylation and suppressing the spread of tau pathology in vivo [82]. With a decrease in serum level in AD patients [83], sirtuin 1 activation could be critical for the reversal of tau accumulation and important for future AD therapies.
Normally, tau is, to some extent, phosphorylated in fetal and adult brains but is not accompanied by the aggregation of tau into filaments. When recombinant non-phosphorylated tau is incubated with polyanions, it is prone to form filaments [84]. This suggests that, except for phosphorylation, unknown mechanisms may participate in pathological filament formation. Under physiological conditions, tau is not prone to polymerize into PHFs, but the process can be accelerated when tau fragments are added that contain only the repeated domain (RD), without the N- and C-terminal domains [85].
Physiological or pathological tau species include: monomers, dimers/trimers, small soluble oligomers, insoluble granular tau oligomers, filaments, pretangles, large nonfibrillar tau aggregates, and NFTs, as well as ghost tangles (the extracellular filaments remaining after the death of tangle-bearing cells) [86, 87]. The formation of tau tangles, generally, involves the following steps: (1) acquiring aggregation-competent conformation, (2) forming dimers and small soluble oligomers (pre-tangles), and (3) generation of filamentous inclusions. There is an ongoing debate as to whether and which forms of tau species are toxic [88]. However, increasing evidence suggests that, of all the tau protein species, the large insoluble inclusions are not the most toxic form; in contrast, oligomeric tau has been identified as an acutely toxic tau species in vivo [89] and in vitro [90] that induces neurodegeneration.
Through interactions with tau, the formation of tau aggregation is negatively or positively regulated by many protein molecules, including the heat shock cognate protein 70 (Hsc70) [91] and the 90 kDa heat shock protein (HSP90) [92], immunophilins FK506 binding protein 51 (FKBP51) and FK506 binding protein 52 (FKBP52) [93, 94], Ras GTPase activating protein binding protein 2 (G3BP2) [95], and Tripartite motif containing protein 11 (TRIM11). For example, FKBP52 exists abundantly in the nervous system. Its decrease in AD brains is closely associated with the accumulation and aggregation of tau [96]. In vitro, FKBP52 directly promotes oligomerization of truncated or mutated tau [97]. Deciphering the implications of FKBPs in neuronal pathology could make them therapeutic targets for AD linked to pathological protein aggregation [98]. Recently, a new endogenous inhibitor of tau protein aggregation (TRIM11) was identified [99]. TRIM11 is a member of the tripartite motif (TRIM) protein family and is significantly down-regulated in AD brains. TRIM11 promotes proteasomal degradation of mutant and excess normal tau, improves tau solubility by serving as a molecular chaperone that prevents tau misfolding, and is a depolymerizing enzyme that disassembles preformed tau protofibrils. TRIM11 was shown to attenuate neuroinflammation, tau pathology, and cognitive impairment through intracranial delivery in animal models of tauopathies, suggesting that restoring TRIM11 expression may represent an effective therapeutic strategy [99, 100].
In AD patients, NFTs occur first in the transentorhinal or entorhinal cortex in the medial temporal lobe (Braak stages I–II), thereafter gradually progress to the hippocampal region (stages III–IV), and finally involve the association neocortex or the primary areas of the neocortex (stages V–VI). In vitro and in vivo evidence has shown that tau pathology appears to propagate in a progressive and stereotypical manner in which misfolded hyperphosphorylated tau first accumulates at the locus coeruleus, from where it spreads to the entorhinal cortex, hippocampus, and neocortex; this process is known as tau propagation and was first postulated in 2009 [101].
In its rudimentary form, tau serves as a ‘seed’ that is introduced or formed, which is transferred to neighboring cells, where it further aggregates in a template-dependent manner. Tau aggregates are capable of transferring from one cell to another and trigger templated misfolding and aggregation of normal tau in healthy cells, thus spreading tau pathology across different brain regions in a prion-like manner. The prion-like propagation of tau follows the following steps: (1) generation of seeding tau capable of propagating the pathology, (2) secretion of seeding tau in a diseased cell, (3) uptake of seeding tau by a neighboring cell, and (4) generation of new tau seeds based on templated misfolding in the recipient cell.
Although the precise mechanisms of tau propagation have yet to be determined [102], its secretory pathway is unconventional vesicular or non-vesicular mediated [103]. Evidence has shown that tau oligomers can be engulfed through bulk dynamin-dependent endocytosis and transported by the endolysosomal pathway in recipient cells [104], suggesting the possibility of transsynaptic spread of tau pathology in vivo.
Some factors mediate the spreading of tau; this might provide novel potential therapeutic targets to slow the development of AD [105, 106]. Low-density lipoprotein receptor-related protein 1 (LRP1) has the ability to control tau endocytosis and its subsequent spread. Downregulation of LRP1 in an in vivo mouse model of tau spread has been suggested to reduce tau cell-to-cell propagation, indicating that LRP1 could be a key regulator of tau spread and a potential novel target for the treatment of this disease [106, 107]. The transcription factor EB (TFEB) is a master regulator of lysosomal biogenesis and plays an important role in the lysosomal exocytosis of tau species. The expression and activity of TFEB increases in response to tau pathology in the brains of both patients with dementia and transgenic AD-model mice [108]. Evidence suggests that TFEB mediates tau exocytosis and, on the contrary, loss of TFEB leads to aggravated tau pathology and spread [109].
Since A
Tau kinase inhibitors are considered as potentially effective therapeutic agents
for AD based on the robust relationship between tau phosphorylation and its
pathological processes. Targeting GSK3
For p38-MAPK inhibition, a clinical trial from a small number of samples (16 AD patients) found that p38-selective inhibition improved episodic memory in patients with early AD [119]. A phase 2 clinical study reported that 24-week treatment with neflamapimod (40 mg, twice a day), a p38 kinase inhibitor, did not improve episodic memory in patients with mild AD; however, the treatment improved synaptic dysfunction (NCT03402659) [120].
Another modifiable protein kinase is Fyn tyrosine kinase, which is considered a potential therapeutic target as it can phosphorylate tau in the N-terminal domain [121, 122]. Saracatinib (AZD0530), a Fyn inhibitor, has been reported to rescue established memory impairment and loss of synapses in AD mice [123]. It is also believed to be safe and well tolerated, according to a phase 1 clinical trial (NCT01864655) [124].
One tau-targeting approach involves the up-regulation of PP2A activity. Sodium selenate specifically enhances the activity of the PP2A/PR55 heterotrimer, which consists of the regulatory B-subunit PR55 and the form of PP2A that directly dephosphorylates tau and reverses memory deficits in an AD model [125, 126]. Selenium treatment at doses up to 30 mg per day for 24 weeks was shown to be safe and well tolerated in patients in a phase 2a randomized control trial of sodium selenate in mild-to-moderate AD [127]. This treatment delivered selenium to the CNS [128], indicating that it could be a promising therapeutic agent.
Evidence has shown that endogenous tau reductions in adult mice lead to no obvious behavioral or neuroanatomical abnormalities [129], presumably due to compensation for loss of tau function by other microtubule-associated proteins. Small interfering RNA (siRNA) or antisense oligonucleotides (ASOs) can reduce tau expression. DeVos et al. [14] designed human tau-specific ASOs, which inhibited loss of hippocampus volume and neurons, suppressed tau seeding activity, prolonged survival, and rescued nesting deficits in the PS19 tau transgenic mouse model of human tauopathy. A phase 1b clinical trial evaluated the safety, tolerability, pharmacokinetics, and target engagement of tau ASO microbule-associated protein tau (MAPT) RX (MAPTRX) in patients with mild AD, and showed a reduction in the total tau concentration in the CSF and an absence of obvious side effects (NCT03186989) [130].
Immunization therapy for AD was launched in 1999, when active immunization of APP transgenic mice with synthetic antibody polymers was shown to markedly reduce cerebral plaque burden. Immunotherapy for tau pathology has received an increasing amount of attention. Tilavonemab is an immunoglobulin G4 (IgG4) monoclonal antibody capable of binding to the N-terminus of human tau and targeting soluble extracellular tau in the brain. A phase 1, single ascending–dose study (NCT02494024) [131] and a phase 2, randomized, placebo-controlled trial (NCT02880956) have shown the long-term safety but lack of efficacy of tilavonemab in the treatment of patients with early AD [132].
Semorinemab (also called RO7105705, MTAU9937A, or RG6100), another humanized IgG4 monoclonal antibody, has been shown to reduce tau-related toxicity in cultured cells and tau accumulation in a tauopathy mouse model [133]. A phase 1 trial of semorinemab demonstrated dose-dependent target engagement and favorable safety. In a clinical trial with prodromal-to-mild AD patients, semorinemab did not attenuate clinical AD progression during a study period of 73 weeks but demonstrated an acceptable and well-tolerated safety profile (NCT03289143) [134]. Another clinical trial showed that semorinemab had an obvious effect on cognition measured by Alzheimer’s Disease Assessment Scale-Cognitive Subscale-11 (ADAS-Cog11), while this did not improve functional or global outcomes (NCT03828747) [135].
Successful antibodies are expected to bind to key epitopes of tau. Using a transgenic tauopathy rat model, it was suggested that active immunization with a peptide (termed ‘AADvac1’ [136, 137, 138]) led to decreases in hyperphosphorylated tau, decreased levels of tau oligomers, and less cerebral neurofibrillary pathology (see review [139]). AADvac1 was reported to be able to induce high antibody titers in almost all patients while maintaining a favorable safety profile, with a safe, strong, and specific immune response against structural determinants of tau protein, making the vaccine a promising candidate for further development as immunotherapy for AD [138].
Methylthioninium, a diaminophenothiazine, serves as an inhibitor of tau aggregation and has the ability to reduce tau pathology and behavioral deficits in transgenic tauopathy mice [140]. An exploratory double-blind, randomized trial of methylthioninium was conducted in 321 subjects with mild-to-moderate AD (NCT00515333) [141]. After continued treatment for 50 weeks (138 mg/day), benefit was observed on the ADAS-Cog scale in mild-to-moderate subjects. Leuco-methylthioninium bis (hydromethanesulfonate; LMTM) is a stable reduced form that has an inhibitory effect on tau aggregation in vitro and in transgenic AD mice [142, 143]. The result of a 15-month, randomized, controlled double-blind, parallel group trial was negative and did not support LMTM as an additional treatment for patients with mild-to-moderate AD (NCT01689246) [144]. Another study [145], however, did support the notion that LMTM could be effective as monotherapy.
In the search for disease-modifying therapies based on tau pathology in AD, various tau-targeting approaches have been and continue to be explored (current summary as shown in Fig. 1 (Ref. [139])). An O-GlcNAcylation-targeting small-molecule inhibitor (MK-8719, reducing hyperphosphorylation) has demonstrated its ability to decrease tau aggregation [78]; a phase 1 clinical trial in humans has been carried out, but more work is needed [146]. There are numerous therapeutic targets that show promise in animal models of AD, but the overwhelming success in animal experiments has yet to translate into benefits for patients.
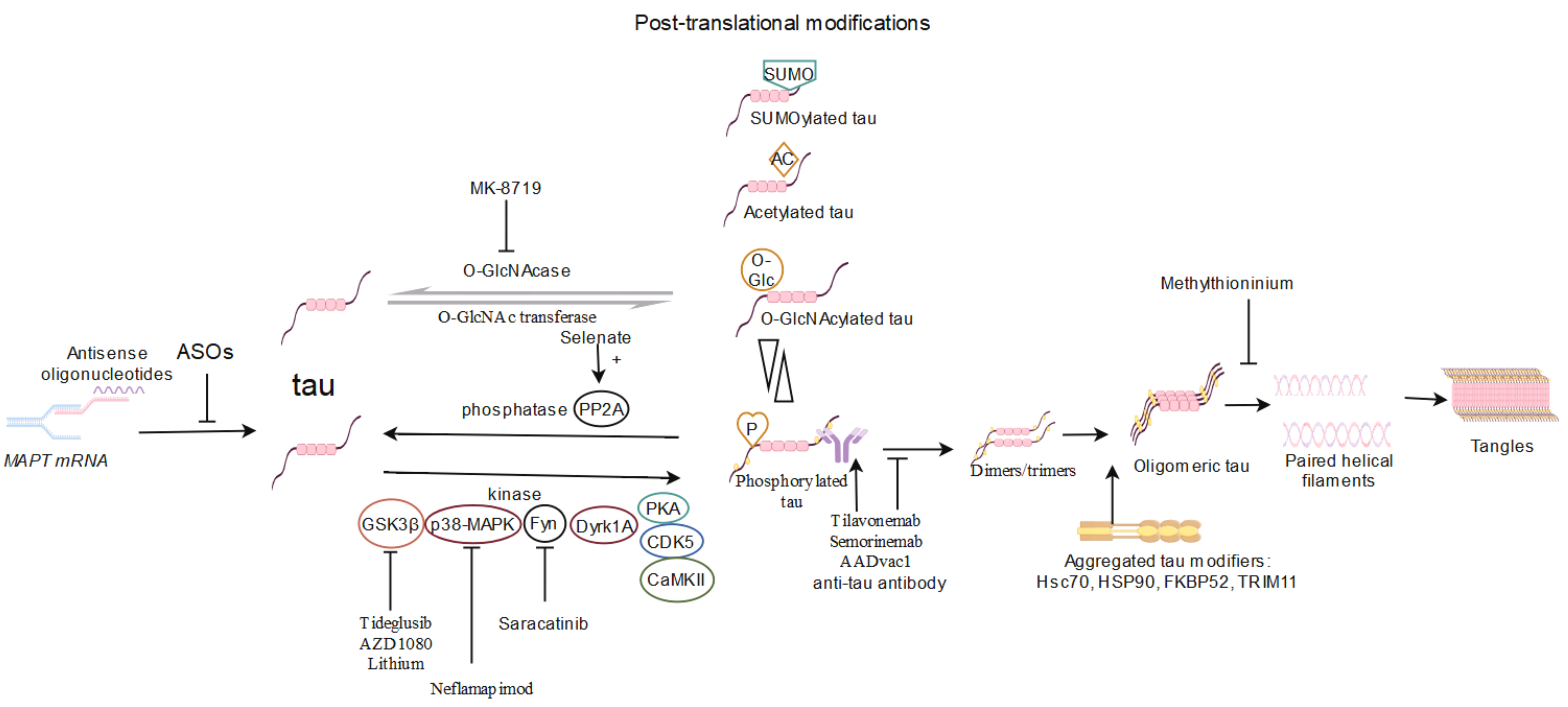
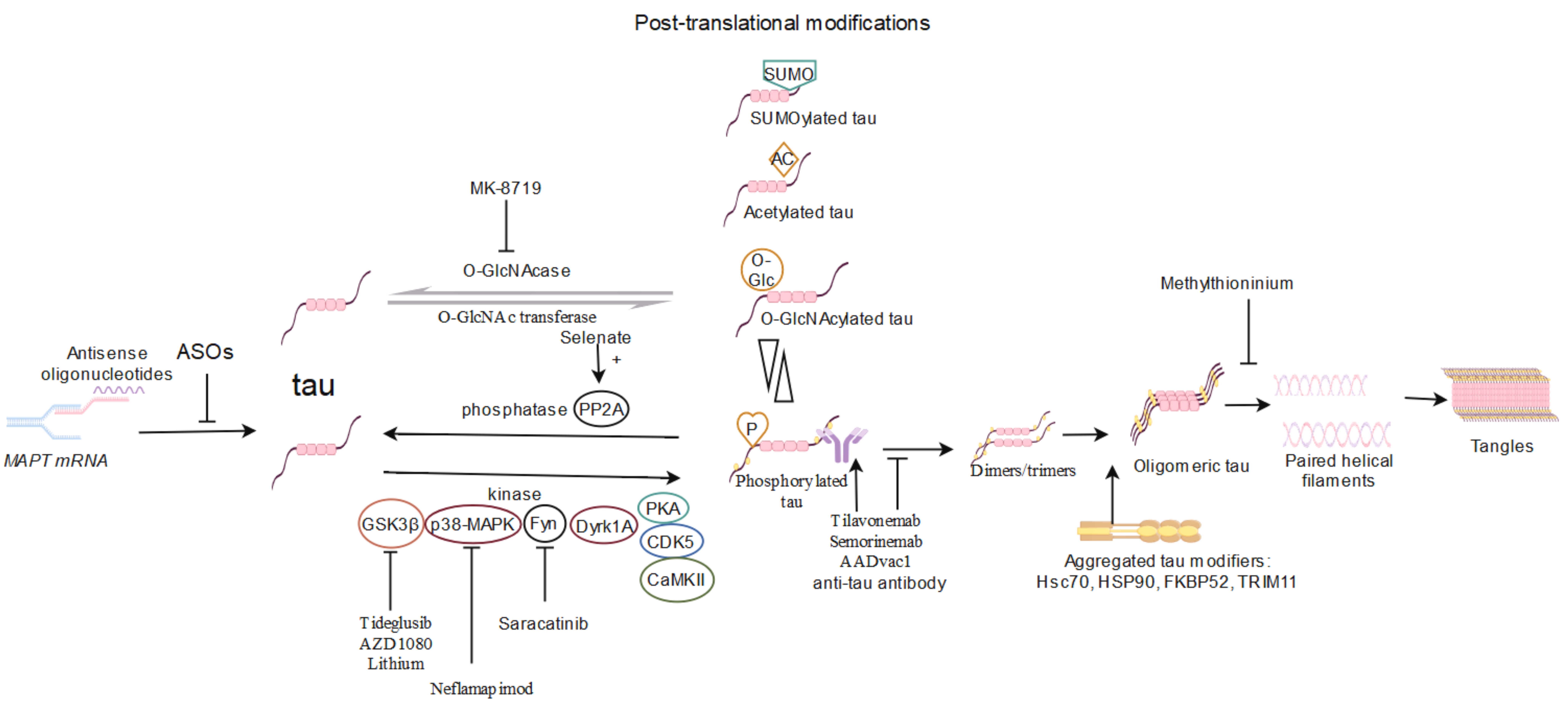 Fig. 1.
Fig. 1.
Overview of tau-targeting therapy based on human clinical trials
in AD. Tau is produced by alternative splicing of MAPT mRNA. Due to the
therapeutic efficacy of reduction of endogenous tau, antisense oligonucleotides
(ASOs) targeting MAPT mRNA have entered clinical trials. In pathological
conditions, tau can undergo post-translational modifications. PTMs include, but
are not limited to, hyperphosphorylation, acetylation, O-GlcNAcylation, and
SUMOylation. Tau is phosphorylated by a series of kinases (GSK3
Initially, potential anti-tau therapies attempted to disrupt toxic gain of function, modulate PTMs, passively eliminate tau, and vaccinate against tau; however, most of these approaches have been discontinued due to toxicity and/or lack of efficacy. Many of the compounds or approaches under investigation might exert effects through one or more pathways; this underlies the complexity of treatment for AD. In fact, immunotherapy remains to be the primary tau-targeting therapy in clinical trials, as it has been shown to be promising in many preclinical studies [136, 139]. Although these multiple therapies have been evaluated as disease-modifying agents in clinical trials, the underlying therapeutic mechanisms remain obscure. In particular, pathogenic tau species are required to be definitively identified in patients with AD. It is not yet known whether a specific oligomeric form, particularly insoluble tau, is a possible source of the key neurotoxic species that can be detected and analyzed in living humans. Achieving this would increase the efficiency of the diagnosis and treatment of AD in future therapies.
In summary, although promising drugs are currently being investigated and there are also more expectations to identify new interventions, more basic research is required to better understand the pathogenesis of AD and decipher the mechanisms of tau dysfunction, to identify more potent disease-modifying therapies.
FZ contributed to the design, summary and writing and presentation of this report. YZ, YS and WC made substantial contributions to the analysis and interpretation of data for partial contents. All the authors had full access to the whole text, and FZ was responsible for the decision of revision and submission. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
The work was supported by the National Natural Science Foundation of China (No. 32160212).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.