
Academic Editor: Beata Sarecka-Hujar
Background: Cardiovascular diseases like stroke cause changes to sphingolipid mediators like sphingosine 1-phosphate (S1P) or its ceramide analogs, which bear the potential to either alleviate or exacerbate the neurological damage. Therefore, the precise identification of alterations within the sphingolipidome during ischemic stroke (IS) and hemorrhagic transformation (HT) harbors a putative therapeutic potential to orchestrate local and systemic immunomodulatory processes. Due to the scarcity of research in this field, we aimed to characterize the sphingolipidome in IS and HT. Methods: C57BL/6 mice underwent middle cerebral artery occlusion (MCAO) and specimens of the peri-infarct tissue were taken for sphingolipid profiling. Results: Ischemic stroke resulted in reduced S1P whilst ceramides were elevated six hours post ischemia onset. However, these differences were nearly revoked at 24 hours post ischemia onset. Moreover, the topmost S1P and ceramide levels were linked to the presence of HT after MCAO. In this study we show the characterization of the sphingolipidomic landscape of the peri-infarct tissue after ischemic stroke and HT. Especially, highest values of S1P, C
Globally, stroke is the second leading cause of death and is a major cause of disability and long-term care. This bears serious socioeconomic consequences for the affected person, their relatives, and society [1]. In 2016, the worldwide prevalence of stroke was 80.1 million, with cerebral ischemia accounting for 84.4% of the cases recorded [2]. Acute ischemic stroke is a medical emergency. The primary goal of ischemic stroke therapy is to achieve a safe, rapid, and effective reperfusion [3, 4]. Current therapeutic approaches are systemic intravenous thrombolysis with recombinant tissue plasminogen activator (rtPA) within a narrow time or endovascular thrombectomy. Although vividly studied more recently, immunomodulatory therapeutic approaches in the acute phase of stroke have yet to demonstrate a benefit regarding clinical outcomes in large scale clinical trials.
Hemorrhagic transformation (HT) represents a complication of ischemic stroke occurring mainly after reperfusion [5, 6]. Thrombolytic therapy (rtPA) increases the risk for HT by approximately 10-fold [7, 8].
However, endovascular mechanical thrombectomy has also been shown to increase the risk of hemorrhagic complications [9]. More than three retrieval attempts during endovascular therapy were associated with an increased rate of symptomatic intracerebral hemorrhage and an Alberta Stroke Program Early CT Score (ASPECTS)
Activation of matrix metalloproteases (MMPs) [12, 13] and severe endothelial damage after ischemia/reperfusion compromise endothelial integrity and foster the development of HT [14, 15]. After transient focal cerebral ischemia, the blood-brain barrier exhibits increased permeability as early as 25 minutes after reperfusion possibly persisting for several weeks [16, 17].
Sphingolipids represent ubiquitous components of cellular membranes involved in cell-cell contacts but also serve as signaling molecules. There is a growing body of evidence regarding their regulatory function following stroke [18, 19, 20]. Ceramides, precursors of the signaling molecule sphingosine 1-phosphate (S1P), play a prominent role as central hubs of the sphingolipid metabolism [21]. As such, they confer and regulate apoptosis [22]. Several studies have provided evidence of ceramide accumulation during cerebral ischemia [23, 24]. The induction of reperfusion is considered to trigger increased ceramide synthesis [25, 26]. Ceramides cause apoptosis through mitochondrial dysfunction [27]. Furthermore, an increase in ceramide synthesis via increased acid sphingomyelinase (ASMase) activity has been demonstrated in animal models of stroke [24, 28, 29, 30]. ASMase-deficient mice exhibited a reduced infarct size and improved neurological deficits after transient focal cerebral ischemia [31]. In contrast, S1P regulates the blood-brain-barrier (BBB) function by conferring its signaling on the vascular endothelium via S1P
In the present study, we aimed at analyzing the sphingolipid metabolome within the peri-infarct cortex following HT. Due to the chemotactic ability of sphingolipids that can regulate local and systemic immunomodulatory processes by recruiting immune cells, the aim of this study was to identify sphingolipid subspecies as putative risk factors for HT after stroke.
For all experiments, male C57BL/6 mice (strain J, 11–12 weeks, Charles River Laboratories, Sulzfeld, Germany) were used and kept on a 12:12 h light-dark cycle with food and water ad libitum. All animal experiments in this study conformed to the German Protection of Animals Act and the guidelines for care and use of laboratory animals as determined by the local institutional review board (Regierungspräsidium Darmstadt, Germany, code FU/1049, approved on 2nd of April 2015). Where experiments required sampling of whole blood isolated from human volunteers, informed consent was obtained.
Transient middle cerebral occlusion (MCAO) was performed as described previously [33]. In brief, mice were anesthetized with 1.5% isoflurane (Forene, Abbott, Wiesbaden, Germany) and received 0.1 mg kg
114 mice were subjected to 3 hours of MCAO followed by immediate imaging and reperfusion. Reperfusion was performed either 6 hours (n = 60) or 24 hours (n = 54) after ischemia onset. Within the 6-hour cohort, 22 mice were treated with warfarin and 38 without, whilst 15 mice received warfarin within the 24-hour cohort (Supplementary Fig. 1). 10 mice were used as sham-operated control. The operations were performed in an unblinded fashion since the operator did not apply any modifications such as drug treatment, however, sample provision for the mass spectrometry or 2,3,5-triphenyltetrazolium chloride (TTC) imaging were done in a blinded fashion.
To increase the chance of spontaneous HT following ischemic stroke (IS) induced by MCAO, we evaluated mice under warfarin treatment at the onset of ischemia versus non-anticoagulated mice. Accordingly, we administered warfarin through drinking water dissolving a 5 mg Coumadin tablet (warfarin sodium crystalline; Bristol Myers Squibb, Munich, Germany) in 375 mL tap water in accordance with previous reports [34]. Assuming a water consumption in rodents of 15 mL/100 g and a body weight of 20 g per 24 hours, a warfarin uptake of 0.033 mg (0.83 mg kg
The neurological examination was performed 6 hours and 24 hours after ischemia onset, respectively. The modified Neurological Severity Score (mNSS) was applied to assess neurological deficits [35]. The mNSS is one of the most frequently referred to neurological scores for the functional assessment of mice after an induced stroke. A scoring system is used to quantify neurological deficits. Hemiparesis, walking behavior, coordination, and sensory function are the main areas assessed. The maximum score for mice is 18 points. The Bederson score was originally developed as an evaluation tool for the success of MCAO in the rat [36]. The neurological deficit is assessed using a 5-point scale and captures behavioral changes in the mouse based on its spontaneous movements. The grip test is used to evaluate motor function as well as coordination. The mice were placed on a wooden bar 30 cm above the ground and the time period to fall off was assessed.
The quantification of sphingoid bases and ceramides in tissue from the peri-infarct cortex collected 6 or 24 hours after ischemia onset as described in detail elsewhere [33, 37]. Briefly, samples of the peri-infarct cortex were snap-frozen with liquid nitrogen and stored at –80 °C until required for further analyses. The tissue samples were first mixed with water:ethanol (75:25, v/v) and homogenized to a suspension of 0.05 mg/
For flow cytometric analysis of peripheral blood immune cells, 1 mL of venous blood after peripheral venipuncture was sampled from a Heparin-lithium monovette. Flow cytometry was performed as described elsewhere [38]. In brief, S1P
The ischemic lesions were visualized by staining murine coronal slices with a 2% solution of 2,3,5-triphenyltetrazoliumchloride (TTC) 6 hours or 24 hours after ischemia onset as described elsewhere [39]. In addition, MCAO mice were assessed by magnet resonance imaging (MRI) 3 hours after reperfusion. MRI measurements were acquired on a 3T Magnetom TRIO (Siemens Medical Solutions, Erlangen, Germany). During MRI, mice were spontaneously breathing after receiving intraperitoneal anesthesia comprising of Ketamine (Ketavet®, 100 mg kg
GraphPad Prism 8 (GraphPad Software, LLC, La Jolla, CA, USA) was used for statistical analyses. Data are illustrated as median
We studied the sphingolipid profile in the acute phase after focal cerebral ischemia, in a model of middle cerebral artery occlusion for 3 hours, followed by reperfusion after 6 hours and 24 hours after ischemia onset. To establish hemorrhagic transformation (HT) more frequently, mice were additionally selected to receive anticoagulation with warfarin (Fig. 1A), which exacerbates the risk of HT [34]. HT was evaluated either using TTC staining or using MRI (Fig. 1B). Mice with effective oral anticoagulation with the vitamin-K-antagonist (VKA) warfarin (0.83 mg kg
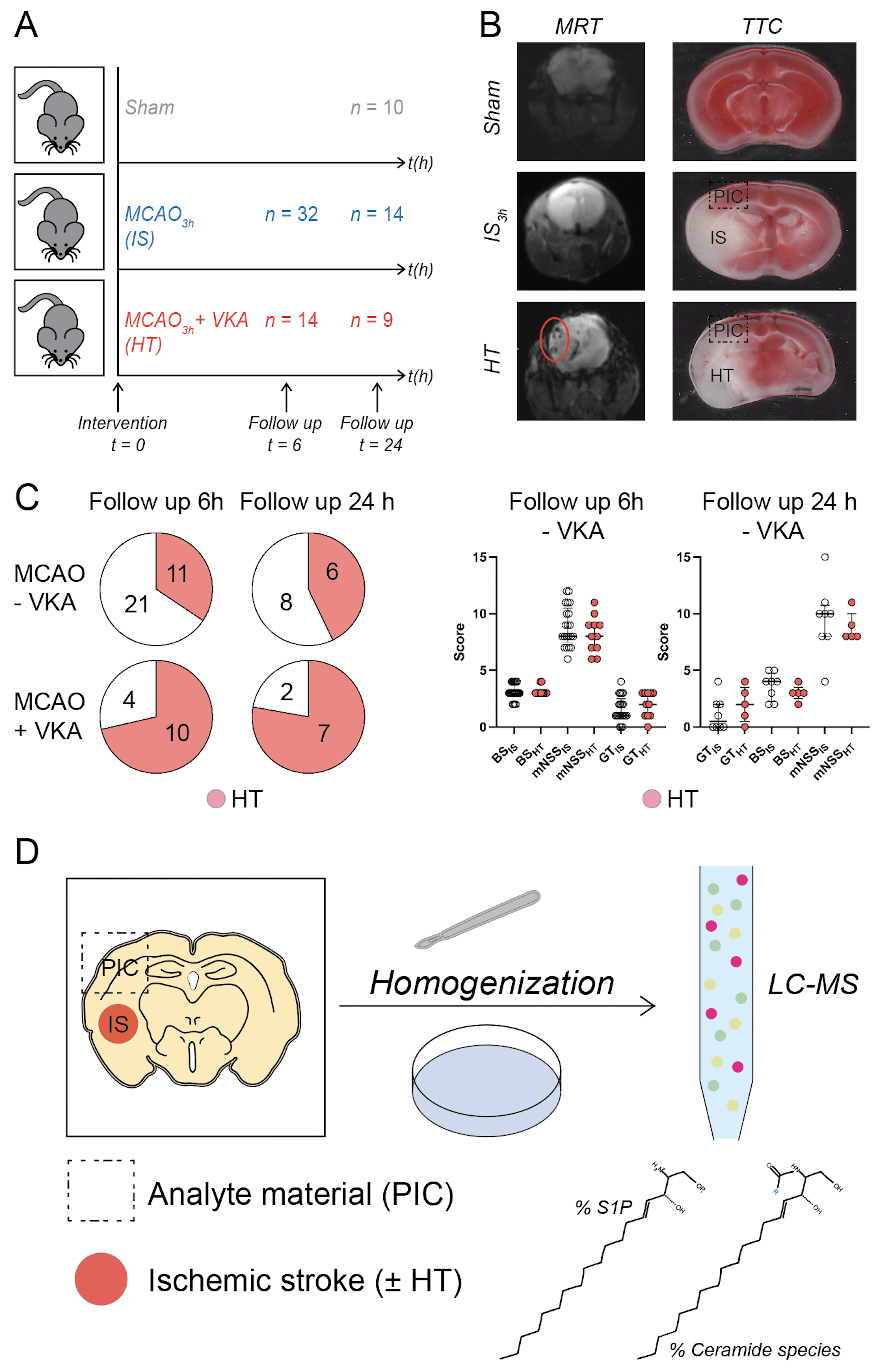 Fig. 1.
Fig. 1.
Peri-infarct cortex tissue sampling to study and the impact of hemorrhagic transformation on sphingolipid mediator concentrations in an unbiased sphingolipid profiling approach. (A) Study design: C57/BL6 mice were either sham-operated (n = 10) or underwent ischemic occlusion of the middle cerebral artery for 3 hours (n = 69) either in the absence (n = 46) or presence (n = 23) of the vitamin-K-antagonist (VKA) warfarin. Mice were observed for 6 hours (n = 46) or 24 hours (n = 23) after ischemia onset. (B) Visualization of large hemispheric ischemic stroke (IS) and hemorrhagic transformation (HT) on TTC staining of native postmortem brain sections and MRI imaging. The peri-infarct cortex (PIC) from which analyte material was taken, is visualized. (C) C57/BL6 mice anticoagulated with VKA displayed hemorrhagic transformation after reperfusion with a higher frequency. In this model of large hemispheric infarctions, HT had no further significant impact on the functional status of the mice. The Mann-Whitney U-test was applied to calculate statistical differences. The data are presented as median
To determine subclinical changes, we next assessed changes on the molecular level in terms of the peri-infarct sphingolipidome by liquid chromatography mass spectrometry (LC-MS) (Fig. 1D). Tissue samples were isolated 6- or 24-hour after the MCAO intervention, homogenized, and analyzed for their sphingolipid metabolome. Results were correlated to the presence of HT that occurred in VKA-anticoagulated but also in non-anticoagulated mice of this study and were recorded for each individual animal.
We could show that in the acute phase after ischemic stroke S1P levels were significantly reduced and ceramides were almost unanimously more abundantly expressed. Indeed, S1P levels were significantly decreased in the periinfarct cortex 6 h after the onset of focal cerebral ischemia in comparison to sham-operated animals (IS
Ceramides, however, displayed a reversed kinetic. For example, C
Owing to the scarcity of analyte material, several special sphingolipidome species were only analyzed in sham-operated mice and such after 24 hours of observation. Here, no differences could be ascertained comparing mice receiving the MCAO intervention with sham (p
 Fig. 2.
Fig. 2.
Reversed kinetics of S1P and ceramide species in the peri-infarct tissue after ischemic stroke. Various sphingolipid species were tested by LC-MS from analyte material sampled from the peri-infarct tissue comparing mice receiving sham-intervention with such having undergone MCAO-intervention and followed up for 6 or 24 hours, respectively. S1P, C
Next, we aimed to characterize the sphingolipidomic changes arising from the establishment of HT after ischemic stroke (IS). Initially, we had observed that HT occurred in a higher frequency if mice had been anticoagulated with VKA prior to the MCAO intervention [34]. Despite some scientific indication that warfarin might affect ceramide expression [40] via inhibition of the production of pro-inflammatory ceramides, we initially treated mice with VKA treatment (rectangles) to enrich for this complication of IS. Indeed, HT (red coloring) did occur more frequently (Fig. 3), but mice also passed away more often (6 h: VKA vs. no VKA: 13.6% vs. 0%, Supplementary Fig. 1). C
 Fig. 3.
Fig. 3.
Gross assessment of the sphingolipidome within the peri-infarct tissue does not distinguish between IS and HT. Cerebral specimens from the peri-infarct cortex of mice that have developed hemorrhagic transformation (HT) of the ischemic core after MCAO were analysed regarding their differential sphingolipid expression profile as compared to mice without HT. A Kruskal-Wallis test with Dunn’s multiple comparisons test was applied to calculate statistical differences. Data are presented as median
Our data demonstrates that HT is not associated with any alterations of the sphingolipidome encompassing the assessment of S1P and various ceramide species neither at 6 hours nor 24 hours after follow up (FU).
S1P can enhance immune cell migration as a chemotactic agent [33, 42] via S1P
 Fig. 4.
Fig. 4.
Chemotactic recruitment of innate immune cells via sphingolipids and in-depth assessment of the sphingolipid subspecies as putative risk factors for HT. (A) As an example of chemotactic recruitment of innate immune cells, whole blood leukocytes were assessed for their expression pattern of the type 1 S1P receptor (S1P
Seeing that S1P is consumed (via receptor binding and subsequent internalization by effector cells) in the acute phase of IS, we intended to risk stratify our established sphingolipidomic profiles by the topmost and bottommost 35% of sphingolipid species measured and qualitative check whether HT had occurred or not (Fig. 4B). In doing so, we identified that apart from C
This study provides comprehensive data on the metabolism of sphingolipid and ceramide species in the peri-infarct cortex after ischemic stroke in conjunction with prior oral anticoagulation therapy and HT.
We report that there is no association in terms of C57BL/6 mice’s neurological performance in the acute to subacute phase (~24 hours) after reperfusion of cerebral ischemia in the context of non-space-occupying HT. Considering the large hemispheric infarctions induced in our experimental paradigm, this is not surprising. In humans, according to ECASS II (European Co-operative Acute Stroke Study II) hemorrhagic events can be distinguished into four subtypes: hemorrhagic infarction (HI) 1 and HI2, and parenchymal hemorrhage (PH) 1 and PH2, respectively [48]. However, further research has shown that predominantly PH2, characterized by
Sphingolipid signaling has emerged as important metabolic pathways in the context of stroke and HT [19, 51, 52, 53]. According to previous studies, we report that ischemia enhances S1P signaling [53]. In an MCAO model based on the inbred mouse strain ICR, the exogenously derived intracerebral application of S1P resulted in an up-regulation of Sphk1 and S1P
We propose that S1P promotes its detrimental consequences by means of intracerebral but also extracerebral signaling. Regarding the latter, we show that S1P
Platelets are another source of S1P release by means of thrombin and factor X activation [66], therefore we sought to investigate the influence of thrombin and factor X on the S1P level [67, 68]. For this reason, vitamin K, the essential factor for prothrombin development, was inhibited by warfarin-supplementation 72 hours prior to assessment. We could not find any impact of VKA-pretreatment on the expression of any sphingolipid nor ceramide species assessed.
Besides S1P and its precursor Spho, our unbiased sphingolipid profiling approach has identified an association of the Hi
In this study, we report changes in the S1P and various ceramide species profile in the peri-infarct cortex associated with HT following IS. Future studies are needed to address the mechanisms within the sphingolipid metabolome by which conversion to these species occurs and which biological consequences are being conferred. Do these species confer a risk for HT or is the conversion of these species a consequence of HT? Do these species adversely affect long-term outcome of the penumbra and survival? What is the mechanistic link to innate immune cell recruitment in terms of chemotaxis, pro-inflammatory cellular activation patterns? Moreover, further experiments are needed to conclusively determine the contributing sources of sphingolipid synthesis and release (e.g., microvascular endothelial cells, neurons, astrocytes, microglia). And considering ongoing trials investigating sphingosine 1-phosphate antagonistic treatment (fingolimod) after stroke, how can long-term immunodepression post-stroke be averted in the context of sphingolipid-antagonistic treatment? Synergistic experiments utilizing intravascular imaging techniques in the context of Tet-On inducible systems could shed some light on these kinds of questions.
AAV, adeno-associated virus; ApoM, apolipoprotein M; ASPECTS, Alberta Stroke Program Early CT Score; BBB, blood-brain barrier; CBF, cerebral blood flow; CD, cluster of differentiation molecule; CNS, central nervous system; FC, fold change; GBD, global burden of disease; HT, hemorrhagic transformation; IQR, interquartile range; IS, ischemic stroke; LC-MS, liquid chromatography mass spectrometry; MCAO, middle cerebral artery occlusion; MMP, matrix metalloprotease; MRI, magnet resonance imaging; PIC, peri-infarct cortex; rtPA, recombinant tissue plasminogen activator; sICH, symptomatic intracerebral hemorrhage; S1P, sphingosine 1-phosphate; S1P
AL, RB, JS designed the research study. AL, ST, DT, KL performed the research. AL, ST, KL, JS analyzed the data. AL and JS wrote the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final version of the manuscript.
The study was conducted according to the guidelines of the Declaration of Helsinki, and all animal experiments conformed to the German Protection of Animals Act and the guidelines for care and use of laboratory animals by the local institutional review board. The study was approved by the Institutional Review Board (Regierungspräsidium Darmstadt, Germany, code FU/1049, approved on 2nd of April 2015). Informed consent was obtained from all subjects involved in the study.
Thanks to all the peer reviewers for their opinions and suggestions.
This research was funded by the German Research Foundation (SFB1039: Project ID 259130777-E02; SFB1039/Z1; SFB1039-TPB08 to R.B). AL is supported as a Clinician Scientist by the SFB1039. JS was funded by a scholarship from the German Academic Scholarship Foundation (Studienstiftung des deutschen Volkes) and by the German Cancer Aid as a fellow of the Mildred Scheel Early Career Center at the CRTD, Medical Faculty, Technische Universität Dresden.
The authors declare no conflict of interest.