
Epilepsy is one of the most common neurology diseases. It is characterized by recurrent, spontaneous seizures and accompanied by various comorbidities which can significantly affect a person’s life. Accumulating evidence indicates an essential pathophysiological role for neuroinflammation in epilepsy, which involves activation of microglia and astrocytes, recruitment of peripheral leukocytes into the central nervous system, and release of some inflammatory mediators, including pro-inflammatory factors and anti-inflammatory cytokines. There is complex crosstalk between the central nervous system and peripheral immune responses associated with the progression of epilepsy. This review provides an update of current knowledge about the contribution of this crosstalk associated with epilepsy. Additionally, how gut microbiota is involved in epilepsy and its possible influence on crosstalk is also discussed. Such recent advances in understanding suggest innovative methods for targeting the molecules correlated with the crosstalk and may provide a better prognosis for patients diagnosed with epilepsy.
Epilepsy, one of the most common neurology diseases, affects nearly 65 million individuals worldwide [1]. It is a clinical entity characterized by recurrent, stereotypical, and spontaneous seizures (a process is known as “epileptogenesis”, which leads to the onset and progression of the disease), followed by various comorbidities that seriously affect a person’s life. Accumulating experimental and clinical data has shown innate and adaptive immunity to be activated and that related inflammatory responses may be induced in epileptogenic foci [2, 3]. The inflammatory response, termed “neuroinflammation”, is restricted to brain resident cells, including neurons, microglia, and astrocytes [2, 4]. Furthermore, it has been suggested that neuroinflammatory signals play a significant role in the progression of epilepsy [5], which involves different conditions, such as release of inflammatory factors (involving pro-inflammatory cytokines and anti-inflammatory factors), activation of microglia and astrocytes, as well as recruitment of peripheral leukocytes into the central nervous system (CNS) [2, 6].
Seizures may induce blood–brain barrier (BBB) leakage [7], which may contribute to crosstalk between resident and peripheral immune responses; specifically, resultant BBB dysfunction may provide a favorable microenvironment for communication between peripheral immune cells (e.g., monocytes and macrophages) and CNS resident immune cells (e.g., microglia and astrocytes), which may be involved in neurogenesis, synaptogenesis, neurotransmission, and angiogenesis after epilepsy or seizures [8, 9, 10, 11, 12].
Importantly, the role of gut microbiota has become an increasing focus in neurological investigations. Lines of evidence have already shown that they are implicated in immunity, inflammation, and central and peripherally associated signaling pathways related to epileptogenesis [13]. Additionally, several lines of evidence have also demonstrated that the composition of gut microbiota can impact not only susceptibility to epilepsy but also its progression [14, 15].
First, evidence originating from studies investigating how the peripheral immune responses and CNS immune responses communicate with each other in epilepsy is reviewed from various perspectives. The goal is to identify novel targets for intervention. Second, the effects of gut microbiota on neuro-immune crosstalk are discussed.
There is growing evidence that monocytes which invade from the peripheral circulation contribute significantly to neuroinflammation and the inevitable consequences that follow a brain insult [16]. For example, in an animal model of encephalitis-induced seizures and hippocampal damage, after the injection of Theiler’s virus into the brain, two groups have separately shown that brain-infiltrated inflammatory monocytes injure the hippocampus [17, 18], which gives rise to the progression of acute seizures [19]. Intriguingly, inhibiting monocyte invasion by injection of clodronate liposomes did not prevent hippocampal damage in a viral encephalitis model [20]. Nevertheless, neuroinflammation accompanied by microglial activation and the infiltration of monocytes was detected in both animal models and patients with temporal lobe epilepsy (TLE) [21, 22, 23]. Taken together, these findings suggest that infiltrating monocytes may in these circumstances be a potential target for preventing or modifying epilepsy.
Macrophages originate from monocytes and enter the CNS via the peripheral circulation after CNS injury [24]. Macrophages can be divided into pro-inflammatory M1 and anti-inflammatory M2 phenotypes [25]. M1 macrophages are neurotoxic, while the M2 subtype enhances axonal regrowth after CNS insult [26, 27, 28]. Interestingly, following spinal cord injury (SCI), a population of macrophages derived from infiltrating monocytes facilitates recovery via the anti-inflammatory cytokine interleukin (IL)-10 [29].
Importantly, chemokines are significant proinflammatory mediators which make
leukocytes penetrate into the brain following seizures. C-C motif chemokine
ligand (CCL)2, alternatively named monocyte chemoattractant protein-1 (MCP-1),
serves as a chemoattractant for the monocyte lineage, which itself is constituted
by microglia, monocytes, and macrophages [30, 31, 32, 33]. The monocyte lineage could
interpret the enhanced expression of monocytes or macrophage in patients with
epilepsy and their animal models. An increased level of CCL2, produced by
macrophages, monocytes or astrocytes, results in disruption of the BBB [34], thus
contributing to epileptogenesis and the onset of epilepsy [35, 36, 37]. With the
exception of CCL2, tumor necrosis factor-
CCR5 and its ligands have further been shown to be involved in vascular
inflammation, while lowered expression of CCR5 may be neuroprotective and lead to
increased neurogenesis [47]. Endogenous astrocytic transforming growth factor
(TGF)-
Lymphocytes infiltrate the CNS accompanied by monocytes, macrophages, and
neutrophils following SE [21, 49, 50], which leads to BBB disruption and
accelerates epileptogenesis [49, 51]. Notably, CD3
A recent experiment showed a biphasic increase of CD45
Furthermore, the epileptic brain was observed to be infiltrated by CD8
Future studies should be conducted to explore specific signaling pathways of peripheral immune cells, especially in the context of refractory epilepsy, to provide further intervention targets originating in the peripheral circulation.
Astrocytes take part in regulating the immune response in the CNS, maintaining
the BBB, secreting cytokines (i.e., chemokines and neurotrophic factors [55, 56])
as well as generating anti-epileptogenic neurosteroids [57]. Astrocytes also take
part in the modulation of inhibitory and excitatory synapses as well as the
mediation of synaptogenesis [58], which significantly contributes to neurogenesis
related to the mediation of the microenvironment [59, 60]. Some reactive
astrocytes have been proposed to re-express factors essential for synaptogenesis
[61] and axonal guidance [62] and possibly contribute to cerebral plasticity and
repair. Reactive astrocytes can be divided into an A1 phenotype (serving a
neurotoxic role) and an A2 phenotype (serving a neuroprotective role) [63].
Disruption of the BBB has been extensively demonstrated both in patients and
animals following acute seizures [35, 49, 64, 65, 66]. Astrocytes in the epileptic
brain also undergo morphological and functional changes, including altered
expression of K
Astrocytes are able to communicate with infiltrated peripheral immune components [55]. While the integrity of the BBB is compromised, peripheral adaptive and innate immune cells, involving neutrophils, monocytes, T-cells, and B-cells, may infiltrate into the CNS, where they mediate different effects including those that are neuroprotective or neurotoxic [16, 22]. Moreover, peripheral leukocytes seem to contribute to the progression of epilepsy by interfering with the BBB [79]. Interestingly, the recruitment of leukocytes is thought to be caused by chemoattractant factors activated by microglia, as well as in response to cytokine signaling [38]. The peripheral leukocytes stick to the endothelial cells of a capillary via adhesion molecules and their relevant ligands and extravasate through the BBB, and produce local inflammation within the BBB. The inflammation contributes to changes in neurotransmission, probably by disrupting the BBB, ultimately leading to the changes in susceptibility to seizure and possible epilepsy [49]. Indeed, several studies have revealed that there is successive recruitment of peripheral immune cells into the CNS after a seizure, paralleled by activation of immune cells in both the periphery and CNS [80, 81]. The mechanism of leukocyte-endothelial interactions and leukocyte trafficking has been confirmed to make an important contribution to the pathogenesis of seizures and epilepsy [82, 83]. A leaky barrier can cause seizures via a positive feedback, thereby promoting the progression of epilepsy. Thus, barrier leakage is not only a result but also a cause of seizures and epilepsy [84].
Astrogliosis is considered to be a protective reaction of astrocytes to inflammation, trauma, pathological neurodegeneration, or ischemic insult [85]. Pro-inflammatory cytokines may greatly influence astrocytes and preserve astrogliosis as well as enhance epileptogenesis [86, 87], which can be generated by activated microglia [88] and reactive astrocytes [89, 90]. Cytokines are involved in both reactive astrogliosis [91] and epilepsy [5]. Chemokine C-X-C ligand (CXCL)1/CXCR2 signaling has been reported to mediate reactive astrogliosis which is closely related to epileptogenesis [92]. Additionally, inhibiting scar formation in STAT3-knockout mice may induce extended lesions, upregulate the loss of neurons and enhance functional deficits after CNS injury, whereas, promoting the formation of scar in mice by a protein suppressor of cytokine signaling 3-knockout exerts the opposite effect [93, 94]. This suggests that astrogliosis and glial scar formation have dual roles and may be neuroprotective in a given context after brain injury [95].
IL-1
In the mouse model of TLE induced bylithium-pilocarpine, specific genetic
elimination of brain-derived neurotrophic factor (BDNF) in astrocytes prevented
an increase in the number of firing neurons; TrkB in astrocytes was genetically
deleted and greatly preserving the spatial learning abilities, which suggested
that the astrocytic BDNF and TrkB molecules serve as promising targets for the
treatment of TLE [99]. The restoration of the normal mode of astroglial
Ca
Microglia may have both pro-epileptic and anti-epileptic effects on the epileptic brain [102]. On the one hand, the activation of microglia may exert an anti-epileptic role by suppressing activity in abnormal neural circuits after SE [103, 104]. On the other hand, chronic activation of microglia may play a pro-epileptic role via inflammatory immune responses [105]. A growing body of evidence has shown that microglial activation can also be beneficial, as it is capable of increasing neurotrophic and anti-inflammatory factors, producing anti-epileptogenic neurosteroids [106], clearing debris, and possibly accelerating repair [107, 108]. Therefore, short-term microglial activation might be favorable [109], whereas, the chronic activation of microglia is possibly detrimental [110] to the development of epilepsy. Reactive microglia have been discovered in animal models of TLE [111] and in surgical samples of patients with epilepsy [112, 113].
Recently, a variety of studies have indicated that activation of microglia
induced by seizures in early life could aggravate susceptibility to seizures in
later life [114, 115, 116]. Also, the activation of microglia in response to seizures
is considered to be the crucial mediator of post-seizure cytokine production
[117, 118, 119]. Microglia are the primary producers of pro-inflammatory cytokines in
response to brain injury [120]. Activated microglial cells can generate cytokines
such as IL-1
Adult seizures may induce a variety of pathological outcomes, including neuronal loss, mossy fiber sprouting, and gliosis and synaptic reorganization in the hippocampus [125, 126, 127]. Microglia can regulate structural and functional alteration of the hippocampus following early-life seizures [128], and microglia can also take part in neurogenesis [129, 130], mediation of axonal processes [131], synapse formation [132, 133], neurotransmitter clearance [134], as well as neuronal phagocytosis [135]. Recent evidence suggests that different phenotypes of microglia may have either positive or negative effects on neurogenesis after brain damage. For example, the M1 phenotype of activated microglia undermines basal neurogenesis [136, 137] and axonal regeneration [138, 139]. On the contrary, the M2 phenotype of activated microglia enhances basal neurogenesis [137, 140] and oligodendrogenesis [137]. The activation state of microglia has been viewed as a key factor in BBB repair and angiogenesis [141] as well as synaptic plasticity after CNS injuries [142, 143]. Therefore, the correct phenotype must be enhanced at the right time to promote appropriate neural repair after CNS damage [144].
A recent experiment revealed that betaine induced microglia to transform into
the M2 phenotype perhaps via hindering TLR4/NF-
Transient receptor potential vanilloid type 1 (TRPV1) is a nonselective cationic
channel that is temperature-sensitive and usually activated by hyperthermia [150, 151]. It is generally suggested that activation of TRPV1 directly facilitates
synaptic transmission and neurogenesis [152, 153]. More interestingly, a recent
study has demonstrated that TRPV1 makes a contribution to the progression of
repetitive hyperthermia-induced seizures (rFS) by blocking the activation of
microglial M2 phenotype via TGF-
Recently, it has been found that myeloid differentiation factor 88 (MyD88) is upregulated in epilepsy models, and that suppression of MyD88 inhibits seizure and neuronal apoptosis [155, 156]. Importantly, inhibition or deficiency of MyD88 has been found to transform microglia and macrophages from the M1 to the M2 phenotype, to finally ameliorate the neurological outcome after SE [157]. Collectively, microglia and their phenotype transformation make an important contribution to the neuro-immune crosstalk between the CNS and the peripheral immune response, an interaction which is likely involved in epileptogenesis.
The role of gut microbiota has become increasingly attractive in recent neurological research. The immune cells within intestinal tissues account for about 70% of the total number of human immune cells [158]. Many reports have shown that interplay exists between gut microbiota and CNS [159], which may influence neuroimmunity. It is known that microbiota and their metabolites are able to affect both human physiology and pathology [160]. For example, the short chain fatty acids (SCFAs), a gut microbiota metabolite, have been demonstrated to insert into the BBB via the bloodstream and directly influence its integrity [161]. The absence of gut microbes induces structural changes in the BBB, these are characterized by reduced tight junction protein levels, consequently enhancing the permeability of the BBB when compared with unaffected mice [162].
SCFAs serve as the necessary metabolites with anti-inflammatory features and decreasing their level may enhance BBB permeability, which consequently enhances neuroinflammation [163]. Intriguingly, administrating sodium butyrate intravenously or intraperitoneally can inhibit BBB breakdown and encourage neurogenesis after traumatic brain injuries [164, 165, 166]. Additionally, treatment with a low dose of penicillin can increase the integrity of the BBB and increase the tight junction density in young mice through long-term alterations in gut microbiota [167].
Recent experiments have demonstrated that gut microbiota is involved in
immunity, inflammation, and both central and peripherally associated signaling
pathways related to epileptogenesis [13]. Indeed, the composition of the gut
microbiota could impact not only human susceptibility to epilepsy but also its
progression [14, 15]. Gut microbiota is regarded as an early biomarker of
epilepsy [168] and there is a strong correlation between the two [169].
Especially with relation to epilepsy, the gut microbiota can change the function
of microglia and astrocytes, the metabolism of carbohydrates and amino acids, the
activity of vagal neuronal activity, and hippocampal neurotransmitter release.
The ketogenic diet (KD) also plays an anti-epileptic role via microbiota [170]. A
KD changes the gut microbiota [171, 172], facilitating selection of microbial
interplays that reduce bacterial
Recent studies have reported that both physical and psychological stress factors have an impact on gut microbiota [175] and it has further been suggested that stress-related changes in gut microbiota may influence the progression of seizures [168]. Recently, it has also been demonstrated that dysbiosis related to chronic stress can promote epileptogenesis, while fecal microbiota transplantation (FMT) from sham-stressed controls transplanted to rats with chronic stress eases the pro-epileptic role of restraint stress [168].
Some experiments have shown that gut microbiota may be involved in both central and peripheral immune growth processes, as well as in the maintenance of the host homeostasis [176, 177]. It has been demonstrated that both a large number of cases of epilepsy preserve an immune-related basis and that immunotherapy is effective in retarding the development of epilepsy [178]. Observational studies have also confirmed that patients with epilepsy may obtain benefits from immunotherapy for controlling seizures [179, 180, 181, 182].
The level of IL-17A has been reported to be greatly upregulated in the
peripheral blood or cerebrospinal fluid (CSF) in those with epilepsy and to be
associated with seizure frequency and severity [183]. The secretion of IL-17 and
IL-6 can be mediated by Bacteroides, and Prevotella may generate great amounts of
SCFAs to take part in regulating cerebral functions [172]. Importantly, symbiotic
gut bacteria have been proposed to modulate Th17 cells [184]. In pediatric
epilepsy and Rasmussen encephalopathy,
There is evidence showing that inadequate neurogenesis can be alleviated by specific strains of probiotic bacteria, hence linking microbiota to hippocampal neuronal regrowth [188, 189].
Also, it is worth noting that recently the function of astrocytes has been found to be influenced by gut microbiota and the cross-talk between them may have great significance in the understanding of CNS diseases [190, 191]. Different kinds of gut bacteria may mediate the astrocytic inflammatory signaling response positively or negatively [191, 192, 193].
Increasingly, methods utilized for changing gut microbiota and reversing dysbiosis are emerging, involved in KD, treatment by probiotics, and as FMT [168, 194, 195, 196, 197].
This review has focused on the crosstalk between CNS immunity and peripheral immunity in epilepsy (Fig. 1), an interaction that possibly provides intervention targets for epilepsy, in particular the specific immune-associated signaling molecules involved in the crosstalk to treat drug-resistant epilepsy. In pathological conditions (i.e., here, epilepsy), peripheral monocytes (or macrophages), T-cells and metabolites of gut microbiota can enter the brain through an impaired BBB and thus interact with central immune components (e.g., microglia, astrocyte). Some possible related pathways may be involved in the crosstalk process, such as the Wingless/integrase-1 (Wnt) signaling pathway, the mammalian target of rapamycin (mTOR) signaling pathway, and zinc signaling [198]. However, the mechanism that describes how and when the peripheral immune components infiltrate and influence the disease (especially during different disease periods) has not been fully elucidated and more study is required.
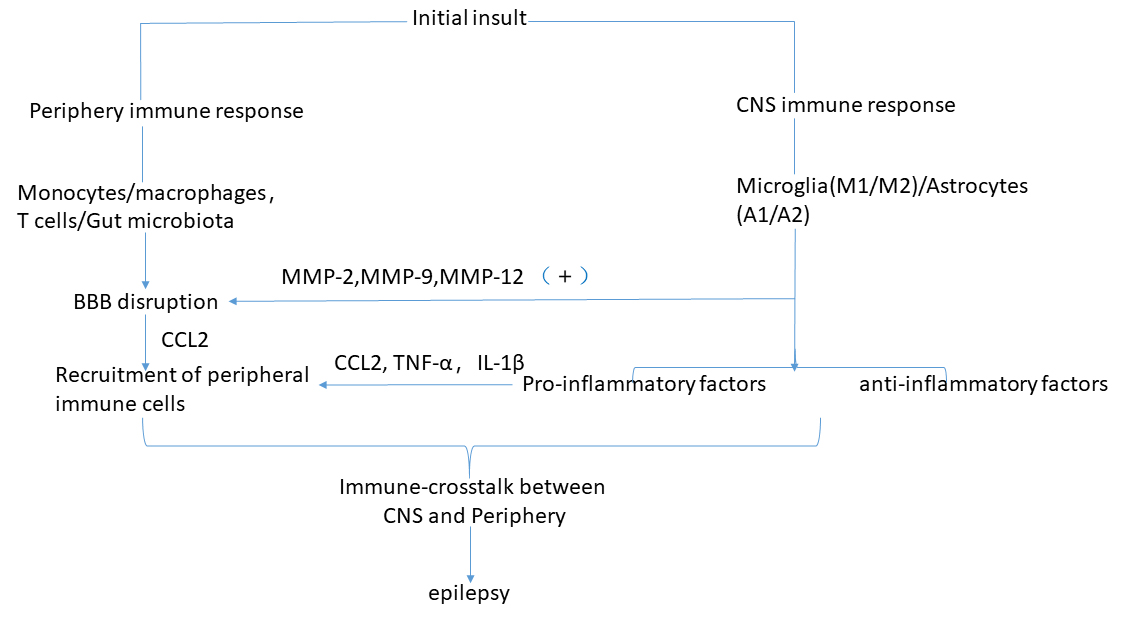 Fig. 1.
Fig. 1.Pathophysiological cascade of events leading from inflammation and microbiota to epilepsy. See Crosstalk between central immunity and peripheral immunity in epilepsy for explanation.
On the basis of recent articles, this review discovered differences in epilepsy and other diseases (such as stroke/amyotrophic lateral sclerosis (ALS)) in the crosstalk between peripheral and brain-resident immune components. For instance, an immune-related crosstalk between the CNS and the periphery has been clarified as beneficial for neuronal repair and functional recovery after a stroke [199]. In the context of ALS, peripheral immune components (e.g., monocytes/macrophages, T-cells) and gut microbiota metabolites can infiltrate into the spinal cord and directly interact with motor neurons (MNs) or surrounding microglia/astrocytes, potentially contributing to either protecting or injuring the MNs as well as resultantly correlating with the survival of patients with ALS [200]. However, crosstalk between the CNS and the periphery in epilepsy mentioned above ultimately contributes to the progression of disease.
Understanding how the crosstalk between the peripheral and the brain-resident immune system influences the initiation and progression of epilepsy may provide a potential approach for clinical treatment. Therefore, it is very important to understand how the peripheral immune components infiltrate into the CNS from the peripheral circulation and intervention at any part of the process may provide interesting and new outlooks into the treatment of epilepsy. Mesenchymal stem cells treatment [201] and immunomodulatory treatment for epilepsy has attracted increasing attention in recent years and needs to be further applied in the clinic. Gut microbiota, especially, play a major role in epilepsy and their alteration or regulation by exogenous intervention may reduce or prevent the disease. A growing number of treatments, containing KD [202], probiotics [203] and FMT [204] have been steadily employed, offering promise for the treatment of epilepsy and its complicated comorbidities. Further studies on gut microbiota and their metabolites in patients with refractory epilepsy will assist in the development of novel intervention targets in the occurrence and progression of the disease.
CNS, central nervous system; BBB, blood brain barrier; TLE, temporal lobe
epilepsy; IL, interleukin; SCI, spinal cord injury; CCL, C-C motif chemokine
ligand; MCP-1, monocyte chemoattractant protein-1; TNF-
XM drafted the paper; XZ have provided assistance in editing and writing the paper; HG, LG, QL and CZ have revised it strictly for important intellectual content. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Not applicable.
We thank the anonymous reviewers for excellent criticism of the article.
This work is supported by the following grants: the Support Plan for Innovation Team of Liaoning Colleges and Universities (No.LT2019015); the Guidance Plan of Liaoning Key Research and Development Plan (No. 2019JH8/10300002); the Support Plan for Cultivate disciplines of China Medical University 2019; the Rejuvenate Liaoning Talents Project; China Medical University High-level Innovation Team Training Plan (No. 2017CXTD02).
The authors declare no conflict of interest.