
Meningiomas are amongst the most commonly encountered intracranial tumors. The majority of these tumors arise intracranially, and the remaining incidents occur along the spinal cord. Meningiomas tend to grow gradually, with many tumors arising in inaccessible locations. Such sporadic behavior poses a therapeutic challenge to clinicians, causing incomplete tumor resections that often lead to recurrence. Therefore, ongoing research seeks to find alternative systematic treatments for meningiomas, with gene-based therapeutics of high interest. Subsequently, genetic studies characterized frequent somatic mutations in NF2, TRAF7, KLF4, AKT1, SMO, and PIK3CA. These genes are communally exhibited in 80% of sporadic meningiomas. In addition, other genes such as the DUSP family, the NR4 family, CMKOR, and FOSL2, have been identified as key players in spinal meningiomas. In this perspective, we aim to investigate current genetic-based studies, with the ongoing research mainly focused on the above NF2, TRAF7, KLF4, AKT1, SMO, and PIK3CA genes and their involved pathways. In addition, this perspective can serve as a potential cornerstone for future genetic analyses of meningioma cases.
Meningiomas derive from the meninges, the tissue layers that cover the brain and spinal cord. With an estimated prevalence of 1.3/100,000 to 7.8/100,000 cases per year as per population-based registries in the US and worldwide, meningiomas constitute the most common intracranial tumors [1]. Approximately 90% of these tumors arise intracranially, while the remaining 10% develop in the spinal cord [2]. Most meningioma cases (80%) are benign and are classified as grade I tumors, as reported in the WHO classification. The other 20%, categorized as grade II and grade III, are considered malignant [3, 4, 5]. Meningiomas constitute around 16% of all intracranial tumors in patients aged between 15 and 39 years, with a female to male ratio of 3.5 : 1 [6]. In the pediatric population, meningiomas usually prompt the suspicion of other NF2 mutation-related diseases such as neurofibromatosis type 2 [7]. Risk factors for meningioma growth are radiotherapy, diabetes mellitus, genetic predisposition, arterial hypertension, hormone intake in women, and possibly smoking [8, 9]. The majority of meningiomas (70–80%) are surgically resected [10]. However, the extent of the resection remains an essential factor of tumor recurrence [11, 12]. Surgically unreachable meningiomas are more prone to inadequate and partial resection and/or may display malignant behavior (WHO grade II and III). As such, these tumors are more likely to grow steadily and recur. For these reasons, treatment remains challenging [13]. Patients with recurring malignancy depleted all conventional therapies—surgical and/or radio-therapeutic—are eligible for systemic therapies [14]. More profound and more extensive knowledge of the genomic foundations in meningiomas is essential for future potential therapeutic targets. Genomic studies have detected that approximately 80% of sporadic meningiomas have recurrent somatic mutations in NF2, TRAF7, KLF4, AKT1, SMO, and PIK3CA[15, 16]. These mutations are associated with the histologic subtype, site, and clinical course of the tumor. Ongoing efforts to molecularly characterize tumor cells are still necessary to establish a better understanding of the epidemiology, prognosis, and potential therapies of the disease.
The first discovery of genetic alterations in meningiomas was in the tumor
suppressor gene NF2, located on chromosome 22 [17]. Loss of this
chromosome is identified in most sporadic meningiomas (40–80%), where almost
60% of these tumors harbor the NF2 gene deficiency, caused by somatic
mutations, allelic loss of chromosome 22, aberrant NF2 promoter methylation, or
epigenetic NF2 inactivation [18]. The loss of the first allele of chromosome 22
induces the loss of the second, leading to the deletion of the entire chromosome
[18]. Although most meningiomas are single and sporadic [19], mutations in the
neurofibromin 2 gene can give rise to the familial syndrome neurofibromatosis 2.
The hallmark of this syndrome is the growth of benign tumors of the nervous
system, namely, schwannomas and meningiomas [20]. NF2 gene produces the
merlin protein, also called Schwannomin, a member of the 4.1 family of proteins,
which act as a bridge between membrane proteins and the cytoskeleton,
cross-linking them. Loss of the merlin protein is characteristic of all NF2
associated meningiomas and nearly half of sporadic cases [21]. Mutations in the
NF2 associated meningiomas often yield a truncated, non-functional
merlin protein, disturbing the normal cellular growth and intracellular
trafficking by disrupting adherent junctions [21, 22]. Recently NF2 was found to
be a negative regulator of the FoxM1/Wnt pathway [23]. NF2 mutations potentiated
by hypermethylation and PRC inhibition are associated with a functional switch
toward the FoxM1 transcriptional program [24]. FoxM1 cooperates with
| Gene | Pathway | Protein product | Function |
| NF2 | Hippo signaling pathway | Neurofibromin 2 (Merlin) | Member of the ERM family of proteins |
| ELK-2 | MAP signaling pathway | Potassium voltage-gated channel subfamily H member 3 | |
| TCI-1 | Rps3-NF-κB-mediated pro-survival pathway | Latexin | Protein inhibitor of zinc-dependent metallocarboxypeptidases |
| KDM5C | Nuclear receptor signaling pathways | Lysine-specific demethylase 5C | Valine, leucine, and isoleucine degradation, hydroxysteroid 17-beta dehydrogenase 10 |
| KDM6A | Transcriptional deregulation in cancer | Lysine demethylase 6A | Histone demethylase. Genetic information processing |
| STMN1 | MAP signaling pathway | Stathmin 1 | Signaling and cellular processes. Environmental Information Processing |
| SMARCB1 | Thermogenesis. Canonical WNT pathway | Subunit of the SWI/SNF complex | SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily B member 1 |
| RIZ1 | Lysine degradation pathway | Retinoblastoma zinc-finger protein | Potential tumor suppressive activities |
| TRAF7 | MAPK and NF-κB pathways | TNF receptor associated factor 7 | Member of the TNF receptor superfamily |
| KLF4 | Signaling pathways regulating pluripotency of stem cells | Krüppel-like factor 4 | Developmental/cell cycle regulator |
| SMO | Hedgehog signaling pathway | Smoothened | Environmental Information Processing |
| AKT1 | PI3K-AKT signaling pathway | RAC-alpha serine/threonine-protein kinase | Regulation of numerous cellular functions |
| ALPL | Folate biosynthesis. Thiamine metabolism pathways | Alkaline phosphatase | Skeletal mineralization |
| *Information retrieved from KEGG Orthology, Uniport and Gene-NCBI. | |||
In meningiomas devoid of NF2 mutation, a notable gene, the TRAF7, was identified [17]. Twenty-five percent of the meningiomas studied revealed TRAF7 mutations [26]. These mutations are also seen in all meningiomas of the secretory subtype [27]. The TRAF7 houses seven WD40 repeats in its carboxyl terminus, with characteristic repeating units of 40–60 variable residues often terminating in a tryptophan and aspartate (W-D) dipeptides [26, 28, 29]. TRAF7 codes for an enzyme known as an E3 ubiquitin ligase, which is involved in multiple signaling pathways including, K3 through its WD40 repeats [17]. Most TRAF7 mutations appear to be linked to the WD40 domains, which participate in the regulation of JNK and p38 MAPK signaling, besides MEKK3, leading eventually to apoptosis [17, 30, 31] (Table 1).
A fraction of NF2 non-mutated meningiomas harbored alterations in AKT1 (~8%), a component of the PI3K-AKT-mTOR pathway [17]. AKT1, a serine/threonine protein kinase, plays a role in activating the PI3K pathway [32]. The AKT1 p.Glu 17 Lys mutation induces constitutive activation of AKT1 protein. Thus, this mutation strongly supports the overactivation of the PI3K signaling pathway in meningioma, which is one of the essential mediators of growth favoring signals [33, 34] (Table 1).
Meningiomas are devoid of AKT1 and NF2 mutations, which constitute between 1% and 5%, harbor mutations in other genes, such as the SMO gene, coding for smoothened homolog protein. This protein belongs to the Hedgehog signaling pathway [16]. The Hedgehog signaling pathway leads to nuclear translocation of protein GLI and activation of target genes associated with cellular growth, proliferation, and angiogenesis [35] (Table 1).
Benign meningiomas (classified as WHO grade I) can potentially harbor recurrent mutations in KLF4. KLF4 is an important transcription factor in oncogenic activation since it intervenes with cell cycle progression following DNA damage. It mediates this process by activating p53-dependent G1/S cell cycle arrest [36]. Mutations in KLF4 have been detected in as many as 50% of NF2-nonmutated meningiomas, but at an interestingly higher rate with TRAF7 mutations, exclusively in secretory meningioma [27] (Table 1).
Around 8% of non-NF2 meningiomas have mutations in epigenetic modifiers such as KDM5C, KDM6A, and SMARCB1 [37]. KDM5C and KDM6A are histone demethylases, while SMARCB1 is a member of the SWI/SNF chromatin-remodeling complex associated with familial forms of meningioma [38, 39]. Several tumors have aberrant histone methylation of the retinoblastoma zinc-finger (RIZ1) methyltransferase gene which is linked to uncontrolled upregulation of the cell cycle [40] since the normal RIZ1 serves as a checkpoint at the G2/M phase of the cell cycle stalling its progression. In meningiomas, the expression of RIZ1 inversely correlates with the tumor grade, where grade I express the highest level of RIZ1 (87.5%), grade II in 38.9%, and grade III in 23.8% of cases [41]. The replacement of RIZ1 expression with a transactivation fusion protein leads to a decline in abnormal cellular growth and proliferation in primary meningiomas and a reduced burden in xenograft mouse models [42]. The research identified a possible role for STMN1 (Stathmin) in the invasion, metastasis, and resistance to drugs of meningioma cells [43]. STMN1 regulates the microtubule system and is involved in specific cell cycle phase checkpoints [44] (Table 1).
The majority of meningioma tumor cells have deletions on Chromosome 22, located at 22q12. This locus is also responsible for making NF2, mentioned above [45, 46]. The second most identified chromosomal abnormality is the deletion of chromosome 1p. FISH studies of 1p may help predict the prognosis of meningioma patients. Genes on 1p are classified as tumor suppressors. Alkaline phosphatase gene (ALPL) is in the deletions’ sites 1p36, 1p35–p32. Its loss was associated with tumor progression [47, 48, 49, 50]. In parallel, loss of heterozygosity of chromosome 14 was identified in 12% to 55% of meningioma cases [51]. Large series of patients identified deletions at the 14q32.2 regions, in which ELK-2, AKT, and TCI-1 are coded [52]. The specific role of these genes is to be studied. Also, deletions on chromosome 10 almost equally occur in meningioma as deletions on 14q [51]. They are attributed to tumor progression rather than formation and are abundantly detected in MII and MIII (40%–50%) than in MI (5%–12%). Loss of heterozygosity at microsatellites predicted higher tumor grades and faster recurrence of the tumor [53].
Spinal meningiomas are the most widely recognized spinal tumors in adults, representing up to 38% of intradural spinal tumors. However, they account for merely 6.5% of total craniospinal tumors in adulthood [54]. Comparable histological subtypes are seen in intracranial and spinal meningiomas, including meningothelial, psammomatous, transitional, atypical, metaplastic, and clear cell types. The former three subtypes are the most widely recognized spinal meningiomas but intriguingly, for unknown reasons, demonstrate a lower hazard for reappearance than their intracranial equivalents [55, 56]. In addition, though cancerous alterations of spinal meningiomas, similar to their intracranial counterparts, are observed, these alterations account for 3% only of reported cases [56]. Additionally, associations were found between several genes and spinal meningiomas: mainly chromosome 22q and its associated gene NF2[57, 58, 59]. Sayagues et al. demonstrated in a microarray-based study of spinal versus intracranial meningiomas, that homozygous deletions on chromosome 22 were more related to spinal rather than intracranial meningiomas [58, 59]. Additionally, spinal meningiomas had a higher likelihood to originate from a single cell clone than from multiple cells [59, 60]. This work also showed a variable expression of 1555 genes in spinal and intracranial meningiomas, among which 35 genes were highly expressed in the spinal counterpart. These genes include Hox genes, the NR4 family of genes, KLF4, FOSL2, and TCF8. Genes involved in intra/extracellular signaling such as RGS16, DUSP5, DUSP1, SOCS3, CMKOR, L6, TGFB1I4, IL1B, CYR61, and CDH2[58, 60] (Fig. 1).
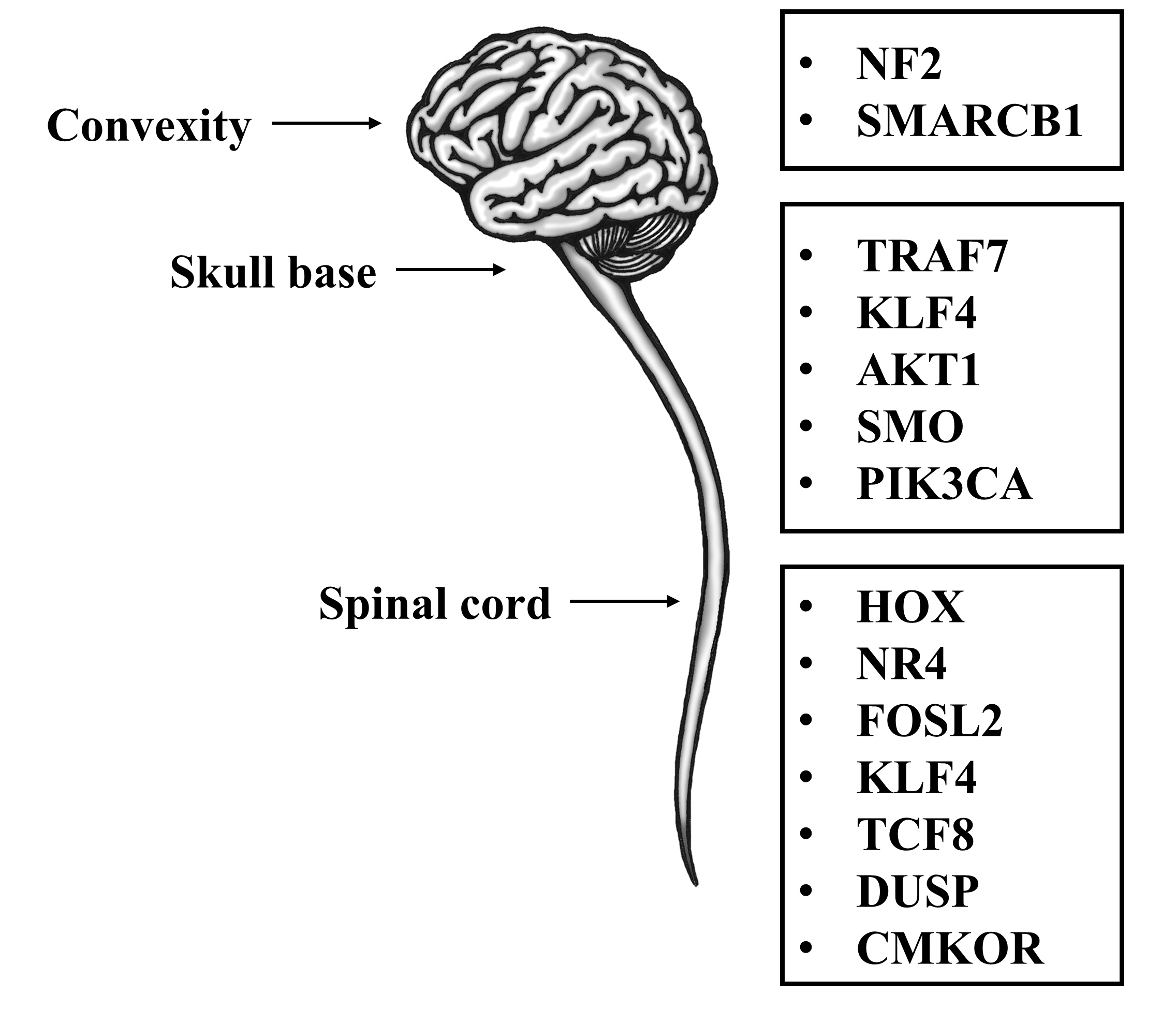 Fig. 1.
Fig. 1.Meningioma tumor locations and their subsequent genetic alterations.
Arslantas et al. [57] histological analyses of 16 patients presented cells from tumors with either complete or partial loss of chromosome 22, with damage of 1p, 9p, and 10q and addition of 5p and 17q, in comparison to chromosomes derived from the patients’ white blood cells. Sayagues et al. [58] also demonstrated that the above 35 genes, while differentially expressed in spinal versus intracranial meningiomas, show no significant association with any of the histological subtypes of meningiomas. Thus, these genes do not explain the predominance of psammomatous, meningothelial, and transitional histological subtypes among spinal meningiomas.
In terms of how spinal and intracranial meningiomas relate, two possibilities are known: (1) The two tumors may correspond to the same tumor type, but at different points in the severity progression, or (2) the two tumors are completely genetically different with shared genetic mutations [58]. The first possibility is supported by the prevalence of chromosome 22 abnormalities among spinal and intracranial meningiomas with intracranial tumors having even more complex and heterogenous abnormalities, which could explain why they have a worse clinical outcome [59]. However, this does not explain the predominance of spinal meningioma in females or its preferred histological subtypes. In addition, many other tumor characteristics remained of higher prevalence among intracranial tumors (proliferative rate, chromosomal abnormality other than with chromosome 22, and tumor cell clones’ number). This supports the possibility that spinal and intracranial meningiomas are different genetically and not just in severity [58]. Table 2 summarizes the genes and pathways that are involved in spinal meningiomas.
| Gene | Pathway | Protein Product | Function |
| Hox | Hox signaling pathway | Hox proteins | Genetic information processing. Transcription factors |
| NR4 | Nicotinate and nicotinamide metabolism | Steroid-thyroid hormone-retinoid receptor superfamily | Nuclear transcription factors |
| KLF4 | Signaling pathways regulating pluripotency of stem cells | Krüppel-like factor 4 | Developmental/cell cycle regulator |
| FOSL2 | Osteoclast differentiation pathway | Fos-related antigen 2 | Genetic information processing. Transcription factors |
| TCF8 | Transcriptional deregulation in cancer | Zinc finger homeobox protein 1 | Genetic information processing |
| RGS16 | Unclassified: signaling and cellular processes | Regulator of G protein signaling 16 | Regulation of G protein signaling |
| DUSP16 | MAPK signaling pathway,Axon regeneration | Dual specificity phosphatase 16 | Dual specificity MAP kinase phosphatase |
| DUSP5 | MAPK signaling pathway | Dual specificity phosphatase 5 | Protein phosphatases and associated proteins |
| DUSP1 | MAPK signaling pathway, Serotonergic synapse | Dual specificity phosphatase 1 | Protein phosphatases and associated proteins |
| SOCS3 | Jak-STAT signaling pathway, TNF signaling pathway | Suppressor of cytokine signaling 3 | Cytokine regulation |
| CMKOR | Cytokine-cytokine receptor interaction pathways | G protein-coupled receptors. Signaling molecules and interaction | |
| L6 | Ribosomal pathway | 60S ribosomal protein L6 | Genetic information processing. Translation |
| TGFB1I4 | TGF-beta signaling pathway | Transforming Growth Factor Beta 1 Induced Transcript 4 | Transcriptional repressor. Acts on the C-type natriuretic peptide (CNP) promoter |
| IL1B | NF-κB signaling pathway, MAPK signaling pathway | Interleukin-1 beta | Mediator of the inflammatory response, cell proliferation, differentiation, and apoptosis |
| CYR61 | AKT/NF-κB signaling pathway | Cysteine-rich angiogenic inducer 61 | Mediates cellular processes. Promotes the adhesion of endothelial cells |
| CDH2 | Cell adhesion molecules (CAMs) | N-cadherin (Cadherin 2) | Calcium-dependent cell adhesion protein. Acts as a regulator of neural stem cells |
| *Information retrieved from KEGG Orthology, Uniport and Gene-NCBI. | |||
In 2015, a clinical trial involving sunitinib, a targeted molecule against vascular endothelial growth factor receptor and platelet-derived growth factor receptor, was one of the first trials to demonstrate a response to targeted therapy on meningioma [61]. Despite not being a gene-targeting therapy, this trial showed promising results and laid the groundwork for other more gene-specific clinical trials. Merlin-deficient meningioma in NF2 mutated cells exhibits constitutive mTORC1 activation and increased growth [62] reported in both the sporadic and hereditary forms of the tumor. When rapamycin’s inhibitory effect on mTORC1 was discovered, numerous clinical trials were conducted to investigate rapamycin analogs in brain tumors [63, 64]. Pachow et al. [65] showed that mouse models that received mTOR inhibitor drugs such as temsirolimus and everolimus had substantially reduced the growth of meningioma cells. Evidence from preclinical studies revealed that vistusertib (AZD2014), a selective inhibitor of mTORC1 and mTORC2, has conceivably more vigorous antiproliferative activity than other synthetic rapamycin analogs. To translate these results into clinical practice, phase II studies enrolling recurrent grade II and III meningiomas are attempting to explore vistusertib effects (NCT03071874, NCT02831257) [66, 67].
In more recent work [68] found that dasatinib, an inhibitor of the EPH RTK/SFK pathways combined with dual mTORC1/2 inhibitor, can lead to even stronger antiproliferative effects than either alone, suggesting that co-targeting both pathways might potentially be an innovative, powerful way to treat patients with NF2-deficient meningiomas. An ongoing phase II trial involving selumetinib, a MEK1/2 inhibitor, studies its effects on NF2 related tumors, including meningiomas (NCT03095248) [69]. Another potential treatment in NF2-associated meningiomas is AR-42, a histone deacetylase inhibitor that arrests the cell cycle and induces apoptosis of the cancerous cells. AR-42 is being evaluated in an early phase I multi-center trial [70]. In progressive/higher-grade meningiomas, promoter region mutations of the telomerase reverse transcriptase (TERT) are linked to worse survival outcomes [71]. This is because mutations in the TERT promoter region entail an increased production of its protein. This type of mutation is classified as the most common single genetic abnormality in Glioblastoma (GBM) [72]. Takahashi et al. [73] demonstrated that eribulin, a microtubule inhibitor, can effectively enter brain tumors and strongly inhibit GBM cells by direct inhibition of RNA-dependent RNA polymerase (RdRP) activity. Another emerging therapy for advanced and aggressive meningiomas is Ribociclib, a cyclin-dependent kinase (CDK) inhibitor. This selective CDK4/6 inhibitor halts the division of malignant cells and is being evaluated in a phase 0/II trial (NCT02933736) [74]. An ongoing clinical trial (NCT02523014) is screening for AKT1, SMO, and NF2 mutations whereby patients receive treatment with corresponding inhibitors [75]. FAK inhibitor (GSK2256098) is among the drugs tested in patients harboring NF2 mutations in this phase II trial. Results published so far yielded significant drug tolerability and better survival rates among patients with recurrent or progressive NF2-mutated meningiomas [76]. Results concerning the response rate and six-month progression-free survival (PFS6) for other targeted genes are still pending. Other inhibitors of the Hedgehog pathway are being tested in trials. Vismodegib, which binds to the SMO receptor and inhibits activation of downstream Hh target genes, was approved for basal cell carcinoma and is under investigation for the treatment of meningioma [77]. Currently, there are ongoing investigations to determine the role of TRAF7 and KLF4 in the development of meningiomas, but to date, there have been no known therapeutic targets for these genetic alterations [26].
Meningiomas, whether intracranial or spinal, are often non-malignant. However, their erratic growth in mostly inaccessible locations poses a challenge for clinicians. In addition, incomplete resection of meningiomas increases the risk of recurrence, prompting researchers to investigate other possible systemic therapies for meningioma treatment. In this perspective, critical genes involved in meningioma have been discussed thoroughly, with 13 unique genes identified for intracranial meningiomas versus 14 genes involved in spinal meningiomas. Amongst these genes, we identified NF2, TRAF7, KLF4, AKT1, the NR4 family, DUSP family, CMKOR, and FOSL2 as significant players in cranial and spinal meningiomas. Many genetic-based therapies against meningiomas are currently being investigated, with various promising results, especially for genes involved in the mTOR pathway. However, more studies are required to unravel the role of other genes involved in meningiomas, such as TRAF7 and KLF4, and their interaction with other known genetic pathways.
ALPL, Alkaline phosphatase gene; KLF4, Kruppel-like factor 4; NF2, Neurofibromin 2; SMO, Smoothened; TRAF7, TNF receptor activated factor 7.
CM and GBM performed a literature search. All authors contributed to manuscript write-up and editorial changes. Finally, all authors read and approved the final manuscript.
Not applicable.
We would like to thank all the peer reviewers and editors for their opinions and suggestions.
This research received no external funding.
The authors declare no conflict of interest.