
† These authors contributed equally.
Regulation of neuroinflammation is critical to control the detrimental impact of chronic stress in the central nervous system. Neuroinflammation occurs in response to chronic stress, leading to enhanced neuronal damage in the brain. We investigated the regulatory effects of stress hormone corticosterone on neuroinflammation regulator, as well as amyloid-β and Beta-secretase 1related signaling. We demonstrate that corticosterone can both positively and negatively regulate amyloid-β expression, which may be related to the ratio of neuroinflammation regulator and Beta-secretase 1 signaling in rat primary cortical neurons. Thirty minutes of treatment with 1μM corticosterone significantly decreased the nuclear translocation of neuroinflammation mediator neuroinflammation regulator (Western Blot: P < 0.05, Immunofluorescence: P < 0.001) and production of Beta-secretase 1 enzyme (P < 0.01), which was accompanied by a reduction in amyloid-β1-42 levels (P < 0.01). In contrast, 1 µM corticosterone treatment over 3 days increased nuclear neuroinflammation regulator localization (P < 0.001), followed by the upregulation of Beta-secretase 1 (P < 0.01) and amyloid-β1-42 (P < 0.05) expression. This work is the first to demonstrate that the duration of corticosterone exposure can promote or inhibit amyloid-β production, and to link this effect with Beta-secretase 1 / neuroinflammation regulator signaling, together with providing valuable insight into the mechanisms of neuroinflammation and neuroprotection.
The stress hormone corticosterone (CORT) is released and regulated in both acute and chronic stress via activation of membrane mineralocorticoid and glucocorticoid receptors (mMRs and mGRs, respectively). CORT participates in the complex regulation of multiple cellular processes related to synaptic function, neuroinflammatory priming, apoptosis, autophagy, and neuroprotection in the central nervous system (CNS) (Frank et al., 2019; Jung et al., 2019; Prager and Johnson, 2009; van Galen et al., 2010). Crucially, neuroinflammation is strongly implicated in neuronal damage, which results from the elevated expression of inflammatory cytokines and chemokines (Kempuraj et al., 2016; Shal et al., 2018).
The involvement of steroid hormones in the neuroimmune system, such as the nuclear factor kappa B (NFκB) signaling pathway, has been widely reported. NFκB is a well-characterized transcription factor comprising combinations of various subunits. These subunits include: RELA (also known as subunit p65), NFκB1 (or subunit p50), NFκB2 (or subunit p52), REL, and RELB. Most NFκB transcriptional activity is regulated by the translocation of a heterodimer (p65/p50) or homodimer (p65/p65) of NFκB subunits into the nucleus, which then activates a series of target genes, such as cytokines, inflammatory enzymes and receptors (Akira and Takeda, 2004; Wang et al., 2015).
As a key contributor to neuroinflammation during stress and injury, the NFκB signaling pathway may play an important role in neuronal damage and neuroprotection (Colombo et al., 2014; Monje et al., 2003; Wang et al., 2015). Interestingly, several studies have reported that the activity of Beta-secretase 1 (BACE1), an enzyme required for amyloid-β (Aβ) production (Yan and Vassar, 2014), is regulated by the NFκB pathway (Buggia-Prevot et al., 2008; Guglielmotto et al., 2012; Wang et al., 2015). Since amyloid plaques (extracellular Aβ deposits formed from amyloid precursor protein (APP)) are key components of the inflammatory response and neurotoxicity (Alasmari et al., 2018; Delgado et al., 2008; Li et al., 2017), it is possible that the activation of NFκB causes increased Aβ production via BACE1.
It was further shown that increased CORT levels, through trans-pineal perfusion of rats with CORT or increased endogenous expression, significantly inhibited nuclear translocation of NFκB (Ferreira et al., 2012; 2005). However, a comprehensive understanding of the effects of CORT on NFκB signaling, and its impact on BACE1 and Aβ expression are still lacking. It is noteworthy that CORT regulates mitochondrial and synaptic function bidirectionally over time (Du et al., 2009; Martin et al., 2009; Popoli et al., 2011). Whether a potential regulatory effect of CORT on the Aβ/BACE1/NFκB signaling also acts in a time-dependent manner remains unknown. In the current study, we evaluated the effects of CORT on the regulation of Aβ1-42 formation, and BACE1/NFκB subunit p65 (NFκBp65) signaling in cultured primary cortical neurons from Sprague-Dawley rats.
Timed pregnant Sprague-Dawley rats were purchased from Kunming Medical University (Kunming, Yunnan, China). All procedures involving animals were in accordance with the Guide for the Care and Use of Laboratory Animals (ISBN 0-309-05377-3) and were approved by the Institutional Animal Care and Use Committee of the School of Medicine, Yunnan University.
Primary cultures of the cells were prepared from 18-day gestation fetuses of Sprague-Dawley rats, as previously described (Du et al., 2009). To prepare cultures of cerebral cortical neurons, cerebral cortical tissues were finely isolated and dissociated using 0.125% trypsin (Gibco, Carlsbad, CA) at 37 ℃ for 15 minutes. After trituration, primary cortical neurons were plated at a density of 4 × 105 cells/well onto plates or coverslips coated with poly-D-lysine (0.1 mg/mL, Sigma). The plating medium [10% fetal bovine serum (Gibco, Carlsbad, CA) in DMEM (Gibco, Carlsbad, CA)] was replaced with fresh serum-free Neurobasal medium (Gibco, Carlsbad, CA) supplemented with B27 (Invitrogen, Carlsbad, CA, USA) and Glutamax (Gibco, Carlsbad, CA, USA) within 24 hours after initial plating, and incubated for 10-12 days at 37 °C and 5% CO2. The serum-free feeding medium was half changed every 3 days.
Serum CORT concentration is approximately 30 ng/mL (86 nM) in unstressed rats, but has been observed to rise to 96 ng/mL (275 nM, upon stress) or 600 ng/mL (1720 nM, upon foot-shock) in stressed rats (Huang et al., 2013; Wichmann et al., 2017). Thus, to mimic physiological and pathophysiological conditions, we treated primary cortical cultures were treated with vehicle (control), low (100 nM) or high (1 μM) doses of CORT (TOCRIS Bioscience, R&D, USA). CORT was delivered by dissolving in culture media for 30 minutes (30-min), 24 hours (24-hour), or 3 consecutive days (3-day).
Nuclear fractions of cortical neurons were prepared using NE-PER™ Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher, USA). According to the protocol supplied by the manufacturer, fresh cells were harvested with a cell scraper in ice-cold PBS and then lysed in Cytoplasmic Extraction Reagent (CER) 1 on ice for 10 minutes. After the addition of CER2, the cells were homogenized and centrifuged at 16000 × g for 5 minutes at 4 °C. The pellet was dissolved in Nuclear Extraction Reagent for 40 minutes on ice and centrifuged at 12000 × g for 5 minutes at 4 °C, collecting the supernatant as the nuclear fraction.
After CORT treatment, the total proteins of cultured cortical neurons were extracted using ice-cold RIPA buffer (Beyotime, Beijing, China) containing Protease Inhibitor (Roche Applied Science, Penzberg, Germany). The lysates were kept on ice for 1 hour and then centrifuged at 14000 × g for 10minutes at 4 °C, and the supernatants were collected containing total proteins. Cellular and nuclear proteins were quantified using a BCA protein assay reagent kit (Pierce, Thermo Fisher, USA). Samples were mixed with SDS loading buffer (Takara Bio, Japan) and boiled for 5 minutes. Equal amounts of proteins were separated on a 10% SDS-PAGE gel and transferred onto PVDF membranes (Millipore, USA). After blocking with 1% BSA (Sangon Biotech, China), the membranes were incubated with primary antibodies against NFκBp65 (1 : 1000, Abcam, UK), histone H3 (1 : 1000, Abcam, UK), BACE1 (1 : 250, Millipore, USA), Aβ1-42 (1 : 500, Cell Signaling Technology, USA) and GAPDH (1 : 5000, Thermo Fisher, USA) overnight at 4 °C. Following washing with TBS-Tween-20 (0.15%), the membranes were incubated with HRP-conjugated donkey anti-rabbit (1 : 1000, Invitrogen, USA) at room temperature for 1 hour. After washing with TBS-T, proteins were visualized using the ECL detection system (Millipore, USA). Images were acquired by the FluorChem E image system (Amersham Imager600, GE, USA), and the relative levels of specific proteins were quantified using Image-Pro Plus Version 6.0 software (Media Cybernetics, USA) and normalized relative to GAPDH or histone H3. Two biological replicates were performed for statistical analysis.
Neurons grown on coverslips were briefly rinsed with PBS, fixed with 4% paraformaldehyde/4% sucrose in PBS for 20 minutes at room temperature, blocking was then performed using 5% normal goat serum in PBST (0.4% Triton X-100 in PBS) for 1 hour at room temperature. The primary antibody, rabbit anti-NFκBp65 (1 : 100, Abcam, UK), was applied for 48 hours at 4 °C. After washing with PBST, the cells were reacted with donkey anti-rabbit IgG conjugated to Alexa Fluor 594 (1 : 300, Jackson Labs, USA) for 90 minutes at room temperature. Coverslips were mounted with an anti-fade mounting medium containing DAPI (Beyotime, Beijing, China) and imaged immediately on a laser-scanning confocal microscope (FV 1200; Olympus; Tokyo, Japan). Five or more images for each coverslip were captured randomly under the same conditions, including laser output strength, exposure time, gain, offset, etc. To investigate the nuclear translocation of NFκBp65, the co-localization coefficient of NFκBp65 and DAPI was quantified using Image-Pro Plus software (Media Cybernetics, USA).
The cell culture plates in this study were randomized into CORT treatment groups and control groups, and the results were analyzed based on a double-blinded method. All quantitative data were combined from 4-10 independent experiments and presented as the mean±SEM (specific n values are indicated). Statistical significance was analyzed using Student’s t-test (to compare two groups), or one-way analysis of variance (ANOVA) followed by a multiple comparison posthoc test (to compare multiple groups) (post hoc LSD test for the total protein expression analyses; post hoc Tukey test for nuclear protein expression and colocalization coefficient analyses). P < 0.05 was considered the threshold of statistical significance.
To determine the effect of CORT on Aβ expression, we prepared primary cultures of cerebral cortical neurons to allow CORT treatment in culture. We first studied treated cortical neurons with different CORT concentrations and temporal exposures for 30 minutes, 24 hours, or 3 consecutive days. The levels of Aβ expression after each treatment condition were measured by western blot analysis. The data shown in Figure 1areveals that 30-min incubation with a high dose of CORT caused a remarkable reduction of Aβ expression level (51.8±26.5%, P < 0.01; Fig. 1a). However, after 24 hours of treatment with a high dose of 1 μM CORT, the Aβ expression level significantly increased to 141.3±23.5% (P < 0.05) (Fig. 1b). After incubating for 3 days, a high dose of CORT was found to induce a remarkable increase in Aβ production, which plays an important role in neuronal damage and disease symptoms, compared with the control (160.0±20.2%, P < 0.05; Fig. 1c). These findings indicate that Aβ expression is controlled by the concentration and duration of CORT exposure in primary cortical neurons. Importantly, the ‘low’ dose of 100 nM did not cause any obvious changes in Aβ expression (Fig. 1).
 Figure 1.
Figure 1.The time- and dose-dependent regulation of Aβ1-42 formation in response to CORT stimulation. Primary cortical neurons were incubated with 100 nM or 1 μM CORT for 30 minutes, 24 hours, or 3 days, after which total proteins were extracted, and western blotting was performed using an anti-Aβ1-42 antibody. Data were analyzed by one-way ANOVA and are presented as the mean±SEM (n = 12-30, post hoc LSD test, #P < 0.05, vs. control). (a) Immunoblotting with anti-Aβ1-42 showed a significant reduction in the high-dose CORT groups after 30 minutes. (b, c) Aβ1-42 expression levels were enhanced in the high-dose groups after treatment with CORT for 24 hours or 3 days.
Given that BACE1 is a cleaving enzyme responsible for Aβ production, we next evaluated the expression levels of BACE1 under CORT stimulation. Primary cortical neurons were treated as previously described, and the total cell protein lysate was immunoblotted with an anti-BACE1 antibody. Consistent with Aβ data, BACE1 expression levels also decreased to 52.7±11.7% (P < 0.01) after 30 minutes of treatment (Fig. 2a). After 24-hour treatment, however, BACE1 levels increased to 154.6±4.5% (P < 0.001) in the 1 μM CORT treatment group (Fig. 2b). After treatment for 3 continuous days, both low and high doses of CORT prominently enhanced the BACE1 expression levels (low dose: 130.3±12.8%, P < 0.05; high dose: 158.5±28.2%, P < 0.01; Fig. 2c). Thus, we confirmed that the concentration and duration of CORT exposure control BACE1 expression in cortical neurons.
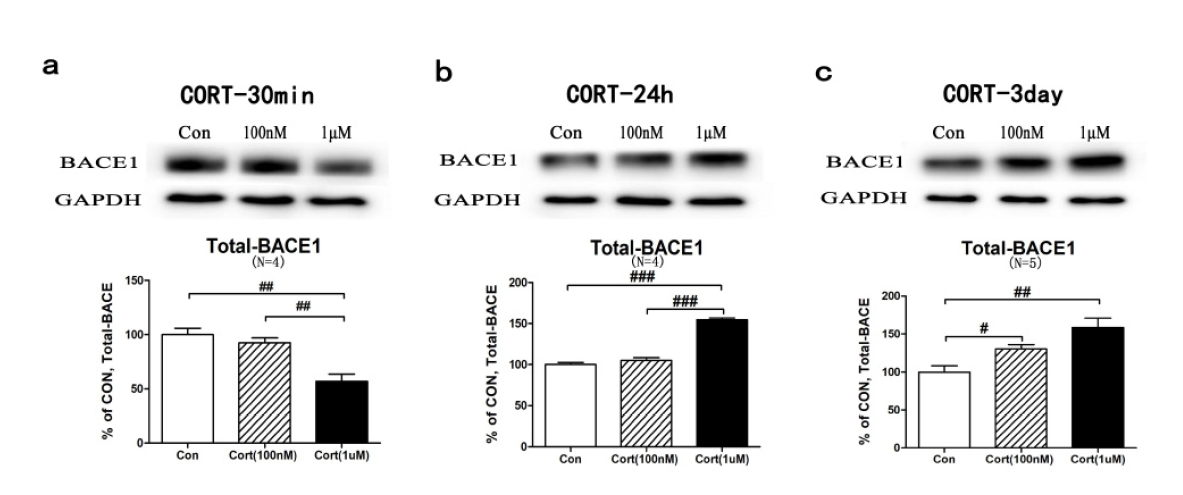 Figure 2.
Figure 2.CORT induced BACE1 expression in a time- and dose-dependent manner. Western blot assays were conducted on the cortical neurons, which were treated with CORT (100 nM or 1 μM) for 30 minutes, 24 hours or 3 days. The whole-cell protein lysates were immunoblotted with an anti-BACE1 antibody. Data were analysed by one-way ANOVA and are presented as the mean±SEM (n = 12-15, post hoc LSD test, #P < 0.05, ##P < 0.01, ###P < 0.001 vs. control). (a) The BACE1 expression levels were downregulated at 30 minutes. (b) The BACE1 expression levels were upregulated at 24 hours in response to CORT. (c) Both a low- and high-dose of CORT elevated the BACE1 levels after 3 days of treatment.
It is well-established that BACE1 expression is modulated by NFκB. To investigate whether CORT impacts the transcriptional activity of NFκBp65, we maintained cortical neurons in culture medium containing 100 nM or 1 μM CORT for 30 minutes, 24 hours, or 3 consecutive days, followed by immunoblotting or immunostaining. As expected, nuclear marker histone H3 levels were significantly enriched in the extracted nuclear fractions (elevated to 391.4±50.4%, P < 0.001; Fig. 3a), as compared with total protein extract. After 30 minutes of treatment, we observed a remarkable reduction of nuclear NFκBp65 expression levels in both low- and high-dose CORT treated groups (low dose: 68.0±19.8%, P < 0.05; high dose: 67.9±18.9%, P < 0.05) compared with that in the control group (Fig. 3b), which is consistent with the findings of BACE1 expression. In contrast, 24 hours of high-dose CORT treatment caused an increase in NFκBp65 nuclear translocation in cortical neuron cultures (202.0±94.3%, P < 0.05; Fig. 3c). After 3 days of CORT stimulation, both low and high doses of CORT elevated NFκBp65 expression levels in the nuclear fraction (low dose: 132.7±38.5%, P < 0.05; high dose: 218.2±41.9%, P < 0.001; Fig. 3d).
 Figure 3.
Figure 3.The dose- and time-dependent curves for the NFκBp65 nuclear translocation induced by CORT. Cortical neurons were treated for 30 minutes, 24 hours, or 3 days, and then the western blot of the nuclear extracts and immunofluorescence staining (IF) of the primary cultured neurons with anti-NFκBp65 (red) antibody and DAPI (blue) were performed. Data were analyzed by Student’s t-test (n = 10, ★★★P < 0.001 vs. total protein) or one-way ANOVA (n = 18-48 for WB, n = 75-105 for IF, post hoc Tukey test, *P < 0.05, **P < 0.01, ***P < 0.001 vs. control), and are presented as the mean±SEM. Purple arrowheads highlight NFκBp65 clusters in the nucleus, while white arrowheads indicate the other. Scale bars, 5 μm. (a) The nuclear marker histone H3 was significantly enriched in the nuclear extracts compared with the whole-cell lysates. (b, e, h) CORT induced a reduction of the NFκBp65 nuclear distribution at 30 minutes, as shown through immunoblot and immunofluorescence assays. (c, f, i) CORT increased the NFκBp65 nuclear translocation in the high-dose group at 24 hours. (d, g, j) Three days of CORT treatment caused an obvious elevation of nuclear NFκBp65 in both the low- and high-dose groups.
To confirm the nuclear distribution of NFκBp65 induced by CORT, we performed immunocytochemistry assays and observed a dose- and time-dependent regulation of NFκBp65 (red) in the primary cortical neurons. Similar to the results from western blot analyses, treatment for 30 minutes significantly decreased the nuclear distribution of NFκBp65 in both the low- and the high-dose CORT treated groups (low dose: 61.2±10.3%, P < 0.01; high dose: 50.2±10.9%, P < 0.001; Fig. 2e and 2h). However, an obvious elevation of NFκBp65 nuclear translocation was observed after 24 hours of high-dose CORT treatment (168.3±23.4%, P < 0.01; Fig. 2f and 2i). Finally, CORT induced a prominent increase in nuclear NFκBp65 after both the low and the high-dose treatment for 3consecutive days (low dose: 223.4±19.0%, P < 0.001; high dose: 263.3±53.6%, P < 0.001; Fig. 2g and 2j). Protein expression levels in all treatment conditions were compared with controls treated with vehicle for corresponding time durations.
We lastly examined the effect of CORT on total NFκBp65 expression in cultured primary cortical neurons (10-12 days in vitro, DIV). Cultured primary cortical neurons were incubated with low (100 nM) or high (1 μM) dose CORT for 30 minutes, 24 hours, or 3 days at 37 ℃, followed by extraction of cellular proteins. The expression level of NFκB subunit p65, a critical component for its transcriptional activity and a contributor to further neuroinflammation, was measured by western blot in cells from each treatment group. The results show that NFκBp65 levels (relative to control) did not change in response to 30 minutes CORT treatment (Fig. 1a), but were significantly downregulated upon high-dose CORT treatment for 24-hour (45.9±9.9%, P < 0.01; Fig. 1b). Intriguingly, after 3-days of treatment, a low dose of CORT decreased, while a high dose increased NFκBp65 expression levels (low dose: 73.8±24.4%, P < 0.05; high dose: 187.8±17.4%, P < 0.001; Fig. 1c), suggesting the time-and dose-dependent regulation of NFκBp65 in response to CORT.
Using a model of primary cerebral cortical cultures from rats, this study found that: 1) 30 minutes’ treatment with high-dose CORT caused a significant reduction in Aβ levels, which was accompanied by markedly reduced nuclear translocation ofNFκBp65 and diminished expression of Aβ producing enzyme, BACE1; 2) 24-hour treatment with high-dose CORT significantly enhanced Aβ1-42 levels as well as BACE1 expression, and NFκBp65 nuclear translocation, but the total NFκBp65 protein level was strongly decreased, likely because of overuse; and lastly, 3) high dose CORT treatment for 3 consecutive days revealed a dramatic increase in Aβ1-42 and BACE1 expression, and in NFκBp65 nuclear translocation. Altogether, these results indicate that the dose and temporal exposure of cells to stress hormone CORT regulates Aβ production and controls BACE1/NFκB signaling activity (Fig. 1-5).
 Figure 4.
Figure 4.The total NFκBp65 expression levels after CORT treatment in cultured primary cortical neurons. Cortical neurons (10-12DIV) were treated with a low (100 nM) and high (1 µM) dose of CORT for 30 minutes, 24 hours or 3 days, and then total cell protein lysates were immunoblotted for NFκBp65. Data were analysed by one-way ANOVA and are presented as the mean±SEM (n = 12-24, post hoc LSD test, #P < 0.05, ##P < 0.01, ###P < 0.001 vs. control). (a) The NFκBp65 remained unchanged after 30 minutes of CORT treatment. (b) The NFκBp65 levels were increased in the high-dose group after 24 hours of treatment. (c) After 3 days of CORT treatment, the NFκBp65 levels decreased in the low-dose group but increased in the high-dose group.
 Figure 5.
Figure 5.A schematic diagram showing the proposed mechanisms through which corticosterone produces beneficial or detrimental effects. (a) A 30-min high-dose of CORT treatment rapidly inhibited NFκBp65 nuclear translocation, which subsequently inhibited BACE1 and Aβ expression. (b) 3-day high-dose CORT treatment significantly induced the upregulation of Aβ expression and the BACE1/NFκBp65 signaling.
Several studies have shown that the glucocorticoid CORT exerts both negative and positive effects on multiple cellular processes depending on its concentration and treatment duration (Du et al., 2009; Harbuz, 2003; Popoli et al., 2011). Here, we focused on the use of cultured primary cortical neurons to investigate the impact of CORT on Aβ formation and the expression of the Aβ producing enzyme, BACE1, to determine its temporal effects. Treatment with a high dose of CORT for 30 minutes significantly inhibited Aβ1-42 (Fig. 1a) expression, BACE1 levels (Fig. 2a), and NFκBp65 nuclear translocation (Fig. 3, b, e, h), suggesting a neuroprotective effect in cortical neurons in response to 30-min corticosterone stimulation. In contrast, 3 consecutive days of exposure to a high dose of CORT enhanced Aβ1-42 expression (Fig. 1c), BACE1 levels (Fig. 2c), and nuclear localization of NFκBp65 (Fig. 3, d, g, and j), indicating the aversive responses involved in neuronal damage.
APP (Amyloid precursor protein) processing comprises two main regulatory mechanisms: the non-amyloidogenic APP cleavage is stimulated by α-secretase, while amyloidogenic pathway regulation occurs via BACE1 activity (Galvao et al., 2019). Multiple studies have shown that the level of BACE1 protein is directly and mostly contributed, if not all, to BACE1 enzyme activity and the formation of its product, Aβ (Lefranc-Jullien et al., 2005; Liebsch et al., 2019; McConlogue et al., 2007; Song et al., 2015; Tallon et al., 2017; Tamagno et al., 2008; Zhang et al., 2007). Thus, our study measured both total BACE1 protein expression and changes in the level of Aβ production. As expected, we observed that30-min treatment with CORT caused a consistent reduction in both BACE1 and Aβ levels, while 3-day CORT treatment elevated the levels of both proteins. It has been well-characterized that by translocating from the cytoplasm to the nucleus, the neuroinflammation mediator NFκB binds to several target genes at NFκB binding motifs and subsequently exert its transcriptional regulation effects (Ferreira et al., 2005). Specifically, binding of NFκB to the BACE1 promoter significantly enhanced the transactivation of the BACE1 promoter, which prominently regulated the expression and activity of BACE1 (Buggia-Prevot et al., 2008; Guglielmotto et al., 2012; Marwarha et al., 2018; Wang et al., 2015). Consistently, we show that 30 minutes CORT exposure to cortical neurons induced a neuroprotective inhibition of NFκBp65 nuclear translocation, which was followed by a reduction of BACE1 expression. However, 3-day CORT exposure showed a detrimental induction of both nuclear translocation of NFκBp65and BACE1 expression, together suggesting a close link between CORT and Aβ/BACE1/NFκBp65 signaling (Fig. 1-4).
The stress hormone CORT is important during acutely stressful situations to cope with dangers in the environment (Du et al., 2009; Prager and Johnson, 2009). Therefore, it enhances synaptic and mitochondrial functions as well as reducing the formation of Aβ. However, when exposed to long-term stress, CORT impairs synaptic and mitochondrial functions and may cause Aβ formation. Emerging evidence indicates that the steady-state balance between the nuclear and cytoplasmic localization of transcription factors is important for transcriptional activity crucial for multiple cellular processes, which allow for adaptation to physical and chemical stresses (Hayden and Ghosh, 2008).This work provides the first functional and systematic evidence that CORT leads to a temporal NFκBp65 nucleus translocation in rat primary cortical neurons, which is followed by regulation of BACE1 and Aβ1-42 (Fig. 5). 30 minutes of exposure to CORT induced a rapid reduction in BACE1 and Aβ1-42 expression, as well as a decrease in nuclear NFκBp65 levels. On the contrary, after a 3-day treatment, CORT caused increase of BACE1 and Aβ1-42 expression as well as increase in NFκBp65 nuclear translocation. These studies provide crucial insight into the cellular responses and mechanisms regulated by CORT in neuroinflammation, neuronal damage, and neuroprotection.
The authors gratefully acknowledge the support of the National Natural Science Foundation of the People’s Republic of China (Grants No.31560274, 31860267, 31650005) and the Academic New Artist of Distinction for Doctoral Post Graduate in Yunnan Province (C176230200).
The authors have no conflicts of interest to disclose, financial or otherwise.