
Human adult stem cells hold promise for regenerative medicine. They are usually expanded for multiple passages in vitro to increase cell yield prior to transplantation. Unfortunately, prolonged culture leads to cell senescence, a major drawback from successful outcomes in cell therapy approaches. Here, we show that an extract from early Zebrafish embryo (ZF1) counteracted senescence progression in human adipose-derived stem cells (hASCs) along multiple culture passages (from the 5th to the 20th). Exposure to ZF1 strongly reduced the expression of senescence marker beta-galactosidase. Both stemness (NANOG, OCT4, and MYC) and anti-senescence (BMI1, and telomerase reverse transcriptase - TERT) related genes were overexpressed at specific experimental points, without recruitment of the cyclin-dependent kinase Inhibitor 2A (CDKN2A, alias p16). Increased telomerase activity was associated with TERT overexpression. Both osteogenic and adipogenic abilities were enhanced. In conclusion, hASCs exposure to ZF1 is a feasible tool to counteract and reverse human stem cell senescence in long-term culturing conditions.
Human resident mesenchymal stem cells (hMSCs) possess self-renewal capacity and multipotency with the ability to differentiate into various lineages (1, 2). They were initially identified in bone marrow (3) and then in various tissues, including the lung, umbilical cord and adipose tissue (4, 5).
During the human life, hMSCs play an important role in replacing old or damaged cells, in order to preserve tissue integrity and oppose senescence-related processes (6-11). The peculiar characteristics of these cells make them amenable in a wide range of cell-based therapies, including clinical applications for many diseases, such as graft-versus-host disease, periodontitis, severe chronic myocardial ischemia, liver cirrhosis, multiple sclerosis and diabetes (2, 12-16). Although hMSCs are present in many tissues, they are few in number. Therefore, cell-based therapy protocols generally require hundreds of million hMSCs per treatment and, consequently, they are subjected to long-term expansion ex vivo, in order to obtain a large amount of cells prior to transplantation (17). Unfortunately stem cells, as well as all cultured primary cells (18), undergo cellular senescence along culture passages, with substantial decline in their differentiation and self-renewal potential (17, 19, 20).
Among hMSCs, those derived from human adipose tissue (hASCs) have been increasingly used as a source for cell-based therapy, due to the ease and minimally invasive technics of tissue harvesting, their robust multipotency and rapid proliferation (2). However, hASCs, as all the other hMSCs, undergo significant senescence. After multiple culture passages in vitro (21-23), as well as in the aging human body (24-26), these cells experience a decline in their repairing capacity, alterations in their differentiating potency (17, 27), and the activation of senescence-related gene expression pathways (17, 27).
Therefore, increased attention has been focused on hMSCs to improve their in vitro expansion in order to counteract, and possibly reverse their natural senescence process. Research is looking for new stimuli to modulate stemness and aging pathways to slow down cellular senescence in culture and for new ways to collect hMSCs. To date, prolonged hMSC lifespan, as well as a reduction of senescent phenotypes, have been obtained with the inhibition of the hystone acetyltransferase (28), the reduction of oxidative stress (29-31), using antagonists of the lysophosphatidic acid (LPA) receptor pathway (32), and culturing hMSCs with rapamycin (33-35). In addition, functional MSCs were isolated as a result from induced Pluripotent Stem Cell (iPS) manipulation, although further researches are necessary to assess both safety and affordability of this method (36-38). Finally, an interesting biophysical approach demonstrated that hASC exposure to electromagnetic fields conveyed by a radio electric asymmetric conveyer (REAC) was effective in counteracting the expression of the senescence marker beta-galactosidase and of senescence-associated gene expression patterning, as well as in regulating stem cell polarity in association with type-2 hyaluronan synthase (39-42).
We have previously described (43) the action of five Zebrafish embryo extracts, collected at different developmental stages (ZF1, ZF2, ZF3, referred to early stages, and ZF4, ZF5 to late stages) on hASCs cultured from the 3th to the 5th passage, providing a preliminary investigation on their effects on cell viability, stemness and senescence regulatory patterning. Late developmental stage extracts decreased cell viability and elicited caspase-3 mediated apoptosis, without significant modification of Bax or Bcl-2 transcription and modulation of stemness and senescence genes. On the contrary, early developmental stages didn’t affect cell viability and apoptosis. Moreover, the early developmental stage ZF1 (5.15 hours post fertilization, hpf), used at a defined dose (10μg/ml) and exposure time (72 hours), showed an ability to modulate stemness gene expression and enhance the transcription of telomerase reverse transcriptase (TERT) and that of the BMI1 proto-oncogene polycomb ring finger (BMI1) gene, involved in anti-senescence pathways (43). These results induced the authors to hypothesize a putative anti-senescence role of ZF1.
This extract was previously analyzed on a one-dimensional Sodium Dodecyl Sulfate - Polyacrylamide Gel Electrophoresis (SDS-PAGE) and its protein content was characterized by using liquid chromatography-tandem mass spectrometry (LC-MS/MS). SDS-PAGE revealed three main protein clusters in ZF1 (44), corresponding to molecular weights of over 45 kDa, around 25-35 kDa and less than 20 kDa, while LC-MS/MS technique allowed to characterize the protein content of the extract. Identified proteins include multiple form of yolk protein vitellogenin, heat shock protein (i.e. HSP8 and HSP70) and other proteins implicated in many pathways as in signaling, cell cycle regulation, protein trafficking, chaperoning, protein synthesis and degradation (44). Moreover, a transcriptomic analysis of the specific developmental stage of our interest (50% epiboly) was previously published to indicate an important transition in gene regulation and transcriptional activity between the 512-cell and 50% epiboly stages (45).
The aim of the present study is to investigate the effect of ZF1 during a long-term hASC culture (until the 20th culture passage), by analyzing the senescence-associated beta-galactosidase (SA beta-gal) activity, the mRNA expression of stemness- and anti-senescence-related genes, as well as the telomerase activity. To gain insights into the biological implication of our observations and assess whether ZF1 may afford a reversion of hASC senescence, we also investigated the potential of this extract for rescuing hASC adipogenic and osteogenic abilities throughout a prolonged culture period.
According to the policies approved by the Institutional Review Boards for Human Studies local ethical committees, all tissue samples were obtained after informed consent (Villalba Hospital Cod. CE: 16076 of Bologna, Italy). Human subcutaneous adipose tissue samples were obtained from lipoaspiration procedures and processed by using the Lipogems device, as previously described in Bianchi et al., 2013 (46). A volume of 1.5 ml of Lipogems product has been seeded in a T75 flask precoated with human fibronectin (0.55μg/cm2) (Sigma-Aldrich Co., St. Louis, MO, USA) and human collagen I-III (0.50μg/cm2) (ABCell-Bio, Paris, France) and cultured in alpha-Minimal Essential Medium (alpha-MEM) (Carlo Erba Reagents, Milano, Italy) supplemented with 10% heat-inactivated Fetal Bovine Serum (FBS) (Gibco, Waltham, Massachusetts, USA), 1% Penicillin-Streptomycin Solution, 1% L-Glutamine 200mM (Carlo Erba Reagents, Milano, Italy), and incubated at 37°C in a humidified atmosphere with 5% CO2 (46). Medium was changed every 4 days and adipocytic Lipogems fraction was removed only after 2 weeks of culture. In our previous study (46), flow cytometry and immunophenotypic analyses, as well as differentiation assays, showed that Lipogems-derived hASCs retained a mesenchymal stem cell identity. Here, Lipogems-derived hASCs were cultured until confluence, then they were harvested by treatment with trypsin-EDTA (Sigma-Aldrich), and subcultured with a medium change twice a week. Experiments were performed on hASCs isolated from three different female subjects (mean age 39 ± 11 years), at subculture passages 5th, 10th, 15th and 20th. All cell cultures were maintained 24h in standard conditions before treatments.
A thousand of Zebrafish embryos at the 50% epiboly (5.15 hpf) developmental stage were harvested and washed in distilled water for 60 sec, as previously described (43, 47). A final preparation containing fertilized eggs at the density of 100/ml of water was turbo-emulsified for 3 min. The final Zebrafish extract (named here ZF1) was resuspended in a glycero-alcoholic solution (60% glycerol, 5% ethanol, 0.12% potassium sorbate and 0.08% sodium benzoate) and stored at 4°C until use, according to the manufacturer’s standard protocol (Aurora Biosearch, Bollate, Milano, Italy). ZF1 was previously analyzed on a one-dimensional Sodium Dodecyl Sulfate - Polyacrylamide Gel Electrophoresis (SDS-PAGE) and its protein content was characterized by using liquid chromatography-tandem mass spectrometry (LC-MS/MS), as detailed in the Introduction (44). ZF1 was previously analyzed on a one-dimensional Sodium Dodecyl Sulfate - Polyacrylamide Gel Electrophoresis (SDS-PAGE) and its protein content was characterized by using liquid chromatography-tandem mass spectrometry (LC-MS/MS) (44). At each experimental time point (subculture passages 5th, 10th, 15th and 20th , respectively) cells were treated with ZF1 10μg/ml for 72h, previously emerged as optimal experimental conditions at early subculture passages (43). An equal amount of ZF1 solvent (a glycero-alcoholic solution) was used as a control in all experiments for 72h (Aurora Biosearch, Bollate, Milano, Italy). In the in vitro toxicology assay (see below), untreated cells were included in the experimental plan.
Protein content of ZF1 was determined with BCA protein assay kit, following the manufacturer’s instructions (Pierce Biotechnology, Rockford, IL, USA). As previously described (43), the protein content of the extract was determined using serial dilution of bovine serum albumin as standard and NanoDrop (NanoDrop ND 1000 v.3.8.1., Wilmington, DE, USA).
The “In vitro toxicology assay kit - Resazurin based” assay (Sigma-Aldrich Co.) was used as a preliminary test on hASCs (culture passage 5th) in order to investigate the effect of ZF1 treatment on metabolic activity of the cells. In this assay, metabolically active cells reduce Resazurin (not-fluorescent and blue) to Resorufin (highly fluorescent and red). hASCs were seeded in quadruplicates in a 96-well plate at 4000 cells/cm2. After 24h in standard conditions, cells were untreated (as control cells) or treated for 72h with ZF1 10μg/ml or with solvent and cultured in complete medium containing 10% Resazurin reagent. In each experiment, negative (blue Resazurin solution with medium) and positive (red totally reduced Resazurin with medium) controls (in absence of cells) were added. Fluorescence (correlated to the presence of reduced Resazurin as marker of cell metabolic activity over time) was measured after 24, 48 and 72h of incubation at 37°C from the beginning of the treatment. The Wallac 1420 Victor2 Multilabel Counter (Perkin Elmer, Waltham, Massachusetts, USA) was used (emission wavelength of 590 nm and excitation wavelength of 560 nm). The number of viable cells correlated with the magnitude of dye reduction was expressed as percentage of Resazurin reduction according to this formula: (FI 590 of test agent - FI 590 of negative control)/(FI 590 of 100% reduced of Resazurin - FI 590 negative control) x 100, where FI means Fluorescence Intensity.
In order to evaluate if the ZF1 treatment had some effect on cell viability, hASCs were seeded at 3500 cells/cm2 in T25 flasks at cultured passages 5th, 10th, 15th and 20th, then incubated for 24h before treatment with 10μg/ml ZF1 or solvent as a control for 72h. Cells were finally detached by trypsin-EDTA, resuspended in complete medium and then counted under a light microscope at least twice with a Neubauer chamber (BRAND GmbH, Wertheim, Germany) in a medium with 50% Eritrosyn B dye 0.2% in PBS (Sigma-Aldrich). Dead cells resulted red. The cell viability was calculated as viable cells counted/total cells counted (viable and dead) x 100. The whole experiment was repeated in cells derived from three different subjects.
Staining was performed using a SA beta-gal Kit (Cell Signaling, Danvers, MA, USA). Briefly, hASCs cultured at passages 5th, 10th, 15th and 20th were treated for 72h with ZF1 10μg/ml or with solvent as control in 6-well plates at the density of 2000 cells/cm2. Each treatment was performed in triplicate, and the whole experiment was repeated in cells derived from three subjects. Subsequently, cells were fixed and then processed according to the manufacturer’s instructions. The number of positive (blue) and negative (not coloured) cells was counted in each sample in at least three random fields under the microscope (at 200x magnification and bright field illumination). The percentage of SA beta-gal-positive cells was calculated as number of positive cells/cell total number x 100 (39).
hASCs were seeded at 3500 cells/cm2 in T25 flasks at cultured passages 5th, 10th, 15th and 20th and incubated for 24h before treatment. Then, cells at specific investigated subculture passages, were treated for 72h with ZF1 at 10μg/ml or with solvent as a control. Each treatment was performed in triplicate at any experimental passage and the whole experiment was repeated in biological triplicates. Total RNA was extracted using the RNeasy Mini Kit (QIAGEN, Valencia, CA, USA) following the manufacturer’s instructions. Genomic DNA contamination was removed by digestion with RNase-free Deoxyribonuclease I (DNase I) (RNase-free DNase set, QIAGEN). The reverse transcription of the extracted RNA was performed as previously described (48) except for the temperature of the reaction that was 37°C instead of 42°C. The success of the reaction was verified by the amplification of the human GAPDH gene, using specific primers (forward sequence: 5’-GAAATCCCATCACCATCTTCCAG-3’ and reverse sequence 5’-GCTACACTGAGCACCAGGTGGTCTCCT-3’). GAPDH amplification was performed as previously described (49) except for 25 cycles instead of 45. Amplicon detection was performed by gel electrophoresis as previously described (43).
Quantitative relative real-time PCR was performed in a Bio-Rad CFX96 real-time thermal cycler (Bio-Rad Laboratories, Hercules, CA, USA) as previously described (50). Briefly, 25ng cDNA were amplified using the SsoAdvanced Universal SYBR Green Supermix (Bio-Rad Laboratories) and 0.2.μM each primer, in technical triplicates for each cDNA sample. The primers were designed by Bio-Rad manufacturer and were specific for NANOG, OCT4, BMI1, MYC, p16 genes (unique assay ID: qHsaCED0023824, qHsaCED0038334, qHsaCED0046537, qHsaCID0012921, qHsaCED0056722 respectively, Bio-Rad Laboratories). Primers for TERT were purchased from Invitrogen (forward sequence: 5’-AAGTTCCTGCACTGGCTGATG-3’ and reverse sequence 5’-GCTTTGCAACTTGCTCCAGAC-3’). Relative gene expression was determined using CFX Manager Software version 3.1. (Bio-Rad Laboratories) with the “delta-delta CT method” (51) and HPRT1, TBP, GAPDH (unique assay ID: qHsaCID0016375, qHsaCID0007122, qHsaCED0038674 respectively, Bio-Rad Laboratories) were used as reference genes. The mRNA levels of hASCs samples treated with ZF1 10μg/ml for 72h were expressed as fold change (2-ΔΔCt), relative to the mRNA levels evaluated in hASCs treated with solvent (control cells) and considered equal to 1. A preliminary gene expression analysis with real-time PCR was conducted to exclude effects of the treatment with solvent.
Telomerase activity was investigated by the aid of TRAPEZE-RT with Amplifluor Primers (Millipore, Bedford, MA, USA). This assay quantifies telomerase activity by measuring real-time fluorescence emission with quantitative PCR. hASCs were seeded at the density of 3500 cells/cm2 in T75 flasks. After 24h in standard conditions, cells were treated with ZF1 at 10μg/ml or with solvent as a control and after 72h, cells were pelleted to perform the assay. The TRAPEZE-RT was performed in triplicate at any experimental passage and the whole experiment was repeated in biological triplicates. Briefly, cells were lysed in 200μl of CHAPS buffer. Aliquots of cell lysate (1μg of protein/well) were assayed in a 96-well quantitative PCR plate. Wells were set aside for generation of the standard curve (TSR8 control template), telomerase-positive cell, Minus Telomerase Control, No Template Control, Heat treated telomerase negative control for each sample and a PCR amplification efficiency control (TSK, K1). Telomerase activity (total product generated) was calculated by comparing the average Ct values from the sample wells against the standard curve generated by the TSR8 control template. Assays were carried out with a CFX-96 quantitative PCR apparatus (Bio-Rad Laboratories).
In order to test the osteogenic potential of hASCs treated with ZF1 and solvent, cells were seeded in a 24-well plate at the density of 9500 cells/cm2. After 72h of treatment, osteogenesis was induced by “StemPro Osteogenesis Differentiation Kit” (Gibco) following the manufacturer’s recommendations as previously described (52). The assays were performed using hASCs at the 5th and 20th culture passages isolated from three subjects. Control cells were incubated with alpha-MEM 10% FBS. After 6 refeeding with osteogenesis induction or alpha-MEM media, extracellular mineral matrix was visible and cells were fixed for 15 minutes with a solution of formalin 4% (Sigma-Aldrich), washed with deionized water (dH2O) and stained with Alizarin Red 1% w/v in dH2O (Sigma-Aldrich) for 10 minutes under mild agitation. Exceeding stain was removed by two washes in dH2O. To perform the quantification, Alizarin Red was solubilized for 15 minutes in agitation using a solution of 10% w/v Cetylpiridinium chloride monohidrate (Sigma-Aldrich) in sodium phosphate dibasic (Sigma-Aldrich; Na2HPO4 10mM, pH=4.1). Absorbances of each sample were read in triplicate at 562 nm using EnSpire Workstation (version 4.13.3005.1482). Data were expressed as the fold change between ZF1 treated cells absorbances and those of solvent treated cells.
In order to test the adipogenic potential of ZF1 and solvent treated hASCs, cells were seeded in a 24-well plate at the density of 10000 cells/cm2. After 72h of treatment, adipogenesis was induced by “StemPro Adipogenesis Differentiation Kit” (Gibco) following the manufacturer’s recommendations as previously described (52). The assays were performed using hASCs at the 5th and 20th culture passages isolated from three subjects. Control cells were incubated with alpha-MEM 10% FBS. After 4-6 refeeding with adipogenesis induction or alpha-MEM media and when lipid vacuoles were detectable, cells were fixed for 15 minutes with a solution of formalin 4% (Sigma-Aldrich), washed with dH2O and stained with Oil Red-O (Sigma-Aldrich; stock solution 0.1% w/v in 2-propanol; stock diluted in dH2O at the ratio 3:2 respectively) for 15 minutes under mild agitation. Exceeding stain was removed by two washes in dH2O. To perform the quantification, Oil Red-O was solubilized for 15 minutes with 2-propanol (Sigma-Aldrich). Absorbances of each sample were measured with the Wallac 1420 Victor2 Multilabel Counter (Perkin Elmer, Waltham, MA, USA) at the wavelength of 490 nm. Data were expressed as the fold change between ZF1 treated cells absorbances and those of solvent treated cells.
Cell viability data and results of osteo- and adipogenic differentiation assays were analyzed using Student’s T-test. Data obtained from SA beta-gal assay were analyzed using Z-test for proportional, while those obtained from real-time PCR were analyzed by the CFX Manager Software version 3.1. (Bio-Rad Laboratories) and by using Wilcoxon test for pairwise comparison. Data from TRAPEZE-RT telomerase activity assay were analyzed by using Wilcoxon test for pairwise comparison. The statistical analysis was performed using SPSS v. 14.0. software (SPSS Inc., IBM, Chicago, IL, USA) and R for statistics 3.2.3. Results were considered statistically significant with p-value < 0.05.
To evaluate the toxicity of solvent and ZF1 on hASCs, an in vitro, Resazurin based, toxicology assay was first performed on cells at culture passage 5th. These results indicated that hASCs treated with both solvent and 10μg/ml ZF1 showed a metabolic activity comparable to that observed in control cells, highlighting the absence of a toxic effect (Figure 1).
 Figure 1.
Figure 1.Effect of ZF1 treatment on hASC metabolic activity. Percentage of reduction of Resazurin in hASCs (culture passage 5th) untreated (CTR) or treated for 72 hours (h) with ZF1 10μg/ml or with solvent (SOLV). Analysis was conducted at 24, 48 and 72h from the beginning of the treatment and data are expressed as mean ± standard deviation (SD).
Then, in order to evaluate cell viability, cell count data were collected from solvent and ZF1 treated samples after 72h of administration. Here, we investigated the viability of hASCs derived from three different subjects in all experimental culture passages (5th, 10th, 15th and 20th). Viability of cells exposed to solvent was comparable with that detected in cells exposed to ZF1: viability data in solvent treated samples spanned from 88% to 95% and those obtained in ZF1 treated cells spanned from 90% to 96%, indicating that ZF1 didn’t significantly affect hASC viability compared with solvent. These results reinforced those obtained with the Resazurin-based assay.
To investigate the ZF1 effect on hASC senescence in a prolonged culture, cells were treated for 72h with ZF1 or with its solvent as a control at culture passages 5th, 10th, 15th and 20th. After treatment, the expression of SA beta-gal was assessed. As shown in Figure 2A, in all investigated culture passages, the number of senescent stained cells was significantly reduced in ZF1 treated hASCs comparing to control cells. Figure 2(B-E) shows representative images of SA beta-gal assay samples, at early and late culture passages (5th and 20th), where it is evident the different number of blue stained cells in ZF1 and solvent treated cells. The observation that even at passage 20th the amount of SA beta-gal expressing cells was remarkably decreased by ZF1 indicates that early developmental stage factors from Zebrafish embryo entailed significant anti-senescence properties.
 Figure 2.
Figure 2.ZF1 counteracts senescence-associated beta-galactosidase activity in hASCs. After overnight culture to allow cell adhesion, hASCs at passages (p) 5th, 10th, 15th and 20th were plated in 6-well plates and were cultured in the presence of ZF1 extract or solvent (SOLV) as a control for 72h, then processed for SA beta-gal assessment. Positive (blue) and negative (not coloured) cells were counted in at least three random fields for each technical replicate under the microscope (at 200x magnification and bright field illumination) and all the ZF1 treated cells were significantly different from the control group (Proportion ± SD; n=3; **p<0.001, *p<0.01) as shown in Figure 2A. Panels B-E show representative images of blue stained SA beta-gal expressing cells, cultured in the presence of solvent (B, D) or ZF1 (C, E) at the 5th (B, C) or 20th (D, E) passage. Scale bar = 100μm.
Preliminary experiments performed with solvent on hASC cultures showed that the glycero-alcoholic solution (the ZF1 solvent) had no effect on stemness gene expression, comparing to control conditions (untreated cells) (data not shown). Analysis of the effect produced by ZF1 on the expression of Nanog homeobox (NANOG), POU domain class 5 homeobox 1 (POU5F1, alias and named in this paper OCT4) and v-myc avian myelocytomatosis viral oncogene homolog (MYC) genes revealed a significant increase in mRNA levels in extract-exposed hASCs, as compared with relative control cells (SOLV), peaking at different time points, according to the investigated transcript. Specifically, NANOG was significantly overexpressed at 5th, 10th and 20th passages (Figure 3A), OCT4 transcription resulted to be increased at 5th, 15th and 20th passage (Figure 3B) and that of MYC was found to be enhanced at the 20th passage in culture (Figure 3C), as showed by fold change (mRNA expression in ZF1 treated cells/ mRNA expression in solvent treated cells, considered as equal to 1).
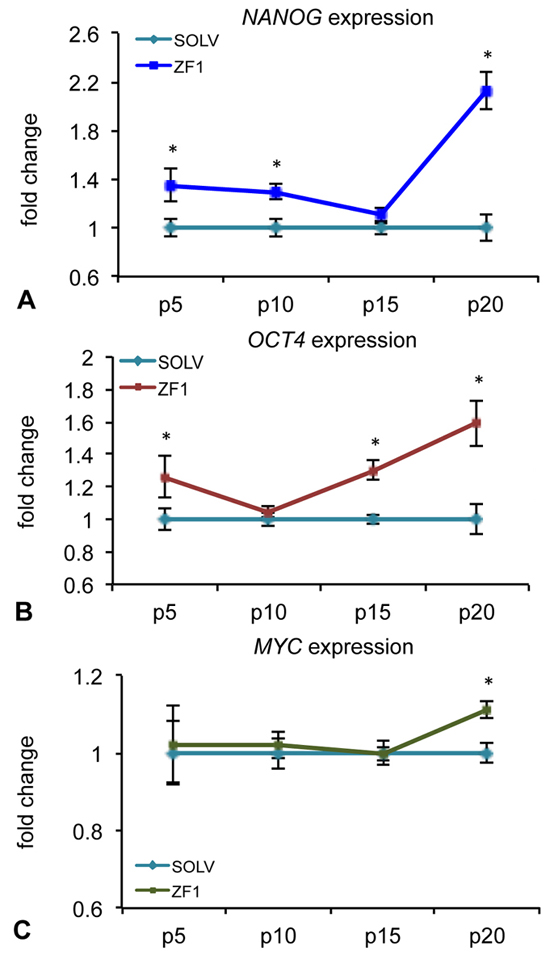 Figure 3.
Figure 3.Effect of ZF1 treatment on NANOG, OCT4 and MYC gene expressions in hASCs. After overnight culture to allow cell adhesion, hASCs at passages (p) 5th, 10th, 15th and 20th were exposed for 72h in presence of ZF1 extract or solvent (SOLV) as a control. The amount of NANOG (A), OCT4 (B), MYC (C) mRNAs expressed in solvent (light blue lines) and ZF1 treated cells (blue, red and green lines, respectively) was normalized to hypoxanthine phosphoribosyl transferase 1 (HPRT1), TATA box binding protein (TBP) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) reference gene expression. The expression of the target genes in ZF1 treated hASCs was plotted as fold change of the expression in control hASCs (considered equal to 1). Statistical significance was evaluated by using Wilcoxon test for pairwise comparison (mean ± standard error of the mean (SEM); n=3; *p<0.05).
Expression of stemness related genes is known to decline after prolonged cell culture in vitro (53). In the developing Zebrafish, pluripotency gene transcription is tightly tuned by factors whose expression is confined within the early developmental stage (45, 54). The present observation that NANOG, OCT4 and MYC gene expression was mainly recovered at late passages by ZF1 indicates that our adult stem cells are able to respond to the early stage developmental factors of Zebrafish with a pluripotency/self-renewal program, and that these factors act when they are mostly needed, in the advanced stage of stem cell senescence.
Preliminary experiments performed with solvent on hASC cultures showed that the glycero-alcoholic solution had no effect on both BMI1 and cyclin-dependent kinase Inhibitor 2A (CDKN2A, alias p16), as well as TERT gene expression, comparing to control conditions (untreated cells) (data not shown). The expression of BMI1, a transcriptional regulator involved in chromatin remodeling and acting as a telomerase-independent repressor of senescence, was significantly increased by treatment with ZF1, as compared with unexposed and relative hASCs, at passages 10th and 20th, as revealed by real-time PCR analysis (Figure 4A). On the contrary, the expression of p16, a cell cycle inhibitor related to an irreversible proliferation arrest, was not affected by ZF1 treatment at any time point (Figure 4B). Exposure of hASCs in the presence of ZF1 for 72h led to an overexpression of TERT, encoding the catalytic subunit of telomerase, compared to relative hASCs treated with solvent, as assessed by real-time PCR, with significance at 5th, 10th and 20th passage of culture (Figure 5A). In addition, after a 72h ZF1 treatment, telomerase activity followed the gene expression trend, with a statistically significant increase in enzyme activity at the 15th and 20th passage in culture (Figure 5B).
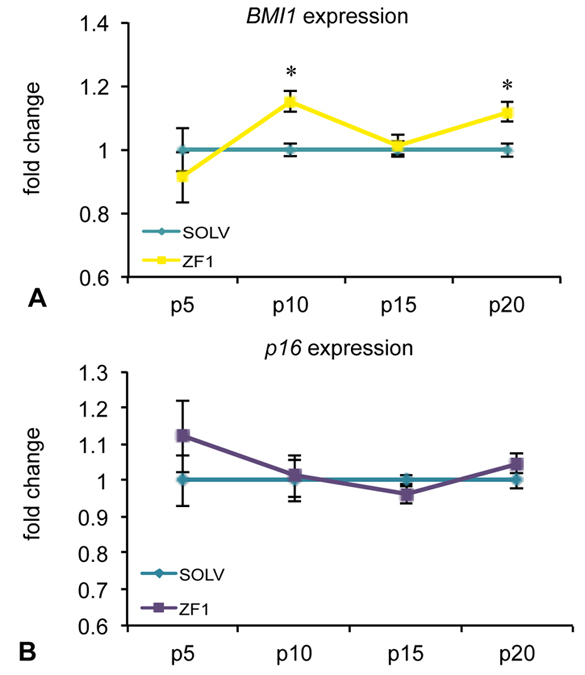 Figure 4.
Figure 4.Effect of ZF1 treatment on BMI1 and p16 gene expressions in hASCs. After overnight culture to allow cell adhesion, hASCs at passages (p) 5th, 10th, 15th and 20th were exposed for 72h in presence of ZF1 extract or solvent (SOLV) as a control. The amount of BMI1 (A) and p16 (B) mRNAs expressed in solvent (light blue line) and ZF1 treated cells (yellow and violet lines, respectively) was normalized to HPRT1, TBP and GAPDH reference gene expression. The expression of the target gene in ZF1 treated hASCs was plotted as fold change of the expression in control hASCs (considered equal to 1). Statistical significance was evaluated by using Wilcoxon test for pairwise comparison (mean ± SEM; n=3; *p<0.05).
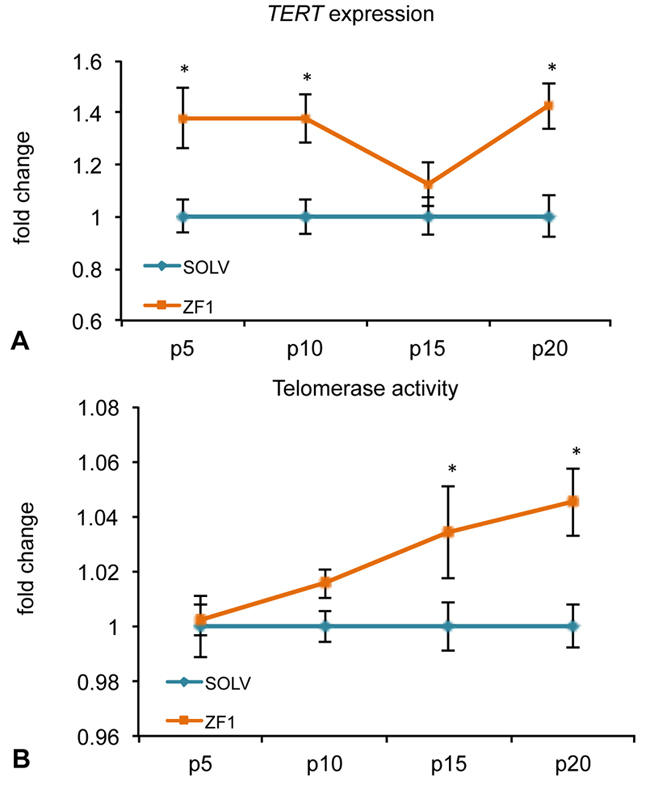 Figure 5.
Figure 5.Effect of ZF1 treatment on TERT gene expression and telomerase activity in hASCs. After overnight culture to allow cell adhesion, hASCs at passages (p) 5th, 10th, 15th and 20th were exposed to ZF1 extract or solvent (SOLV) as a control for 72h. A) The amount of TERT mRNA expressed in solvent (light blue line) and ZF1 treated (orange line) cells was normalized to HPRT1, GAPDH and TBP reference gene expression. The expression of the target gene in ZF1 treated hASCs was plotted as fold change of the expression in control hASCs (considered equal to 1). B) To assess telomerase activity, total cell lysates were analyzed by TRAPEZE-RT assay with Amplifluor Primers. Telomerase activity was calculated by comparing the average Ct values from the ZF1 and control samples against a TSR8 standard curve. Enzyme activity in ZF1 treated hASCs was plotted as fold change of the activity in control hASCs. Statistical significances were evaluated by using Wilcoxon test for pairwise comparison (mean ± SEM; n=3; *p<0.05).
BMI1 and TERT transcription have been implicated in the modulation of telomerase-independent and -dependent pathways, respectively, both counteracting aging processes in vitro and in vivo (41). The increase in BMI1 and TERT gene expression observed here in the presence of ZF1 is consistent with our previous observation limited to the ability of this embryo extract to activate the gene expression of both BMI1 and TERT in hASCs at early (3rd and 5th) culture passages (43). The finding that ZF1 was able to increase BMI1 transcription even at late cultured passages is worthy of consideration, in view of the growing data indicating BMI1 as a major anti-senescence player whose expression is transcriptionally down-regulated when cells undergo replicative senescence (11, 55, 56). At the same time, the observed lack of an effect of ZF1 on p16 transcription at all investigated culture passage suggests that ZF1 action in hASCs may not involve BMI1/p16/pRB pathways (57). Compounding the antisenescent action of ZF1 is its ability to recruit also a telomerase-dependent pathway, as it is inferred by the increase in both TERT gene expression and telomerase activity induced by this embryo extract at the latest investigated passages. In particular, TERT antagonizes cell senescence by opposing telomere shortening. Consonant with this role, a decrease in TERT expression was found to be associated with a decrease in neuroblast growth and differentiation in the developing brain (58).
To evaluate whether ZF1 may have a role in rescuing the differentiating potential of hASCs, an osteogenesis differentiation assay was performed. ZF1 (10µg/ml) significantly enhanced osteogenic differentiation in hASCs when compared with cells treated with solvent as a control, both at early and late culture passages (5th and 20th) (Figure 6). Interestingly, the differentiation ratios (i.e. fold change) were comparable at the 5th and 20th passages (Figure 6A). This finding suggests that the extract could act on osteogenic differentiation pathways, probably by “rejuvenating” the differentiating machinery of hASCs.
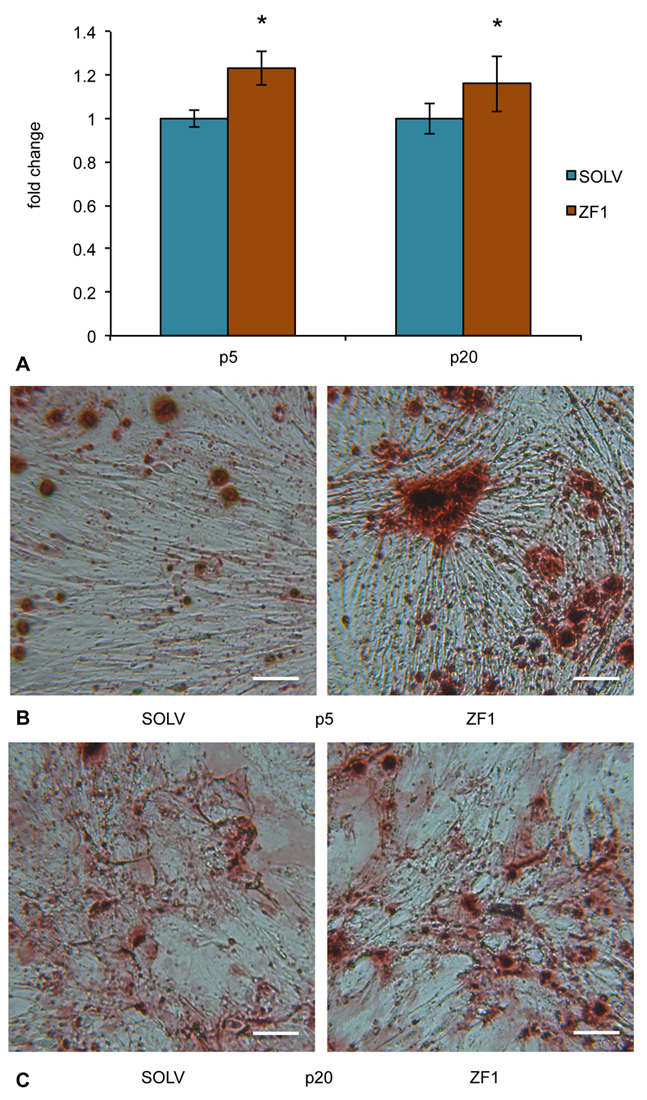 Figure 6.
Figure 6.Effect of ZF1 on osteogenic differentiation of hASCs. After overnight culture to allow cell adhesion, hASCs at passages (p) 5th and 20th were exposed for 72h in presence of ZF1 extract or solvent (SOLV) as a control. A) At the end of the 72h, ZF1 and SOLV treated hASCs were committed to osteogenesis, changing the differentiation medium twice a week for 3 times. Statistical significance was evaluated by using Student’s T-test and data were expressed as fold change in absorbance from ZF1 treated cells, as compared with solvent treated cells (mean ± SEM; n=3; *p<0.05). Panels (B) and (C) show representative images of Alizarin Red staining from SOLV and ZF1 treated hASCs at 5th and 20th culture passages, respectively. Cells positive for osteogenic differentiation showed the formation of mineralized matrix colored in brown/red. Scale bar corresponds to 100μm.
hASC differentiation toward adipogenesis was also evaluated both after chemical induction (namely with StemPro adipogenic induction medium) and in standard conditions of culture, since hASCs after prolonged and overconfluent culture, naturally undergo differentiation. In both experimental conditions, hASCs at the 5th passage differentiate in adipocytes after 72h in presence of ZF1 10µg/ml more efficiently than with solvent (p<0.01) (Figure 7). On the other hand, at the 20th passage, the differentiation ratios (i.e. the fold change) of hASCs subjected to both chemically-induced and standard conditions were weakly increased by ZF1 exposure (Figure 7A and 7C). These observations indicate that ZF1 could act as an enhancer of hASC differentiating potency at early culture passages, without remarkable effects in the regulation of adipogenic pathway at late passage.
 Figure 7.
Figure 7.Effect of ZF1 on adipogenic differentiation of hASCs. After overnight culture to allow cell adhesion, hASCs at passages (p) 5th and 20th were exposed for 72h in presence of ZF1 extract or solvent (SOLV) as a control. At the end of the 72h, ZF1 and SOLV treated hASCs were maintained in standard culture condition (A) or committed to adipogenesis (C), changing the basal or differentiation medium twice a week for 3 times. Statistical significance was evaluated by using Student’s T-test and data were expressed as fold change in absorbance from ZF1 treated cells, as compared with solvent treated cells (mean ± SEM; n=3; **p<0.01). Panels (B) and (D) show representative images of Oil Red-O staining from SOLV and ZF1 treated hASCs that had been maintained at the 5th culture passage under standard (B) or chemically induced (D) conditions, respectively. Cells positive for adipogenesis showed red colored vacuoles in cytoplasm after Oil Red-O staining. Scale bar corresponds to 100μm.
The ability of hASCs to progress along osteogenic and adipogenic lineages is remarkably hampered by donor age (59) and long-term in vitro expansion (17, 60). The observed ZF1 action of increasing the osteogenic commitment at both early and late passages, while failing to sustain adipogenesis in long-term cultures suggests that ZF1 may not play a major role in regulating the adipogenic fate in senescent hASCs. This finding may have remarkable biomedical implication in light of the antithetic execution of adipogenesis and osteogenesis observed in bone marrow MSCs in vitro and in vivo (61, 62). To this end, the effectiveness of ZF1 in reversing the enhancement in adipogenic commitment observed in age-related osteoporosis at the expenses of the osteogenic fate may be exploited in future approaches aimed at counteracting the loss of bone mass in elderly patients, currently a major cause of morbidity and mortality worldwide.
Zebrafish retains considerably higher self-healing properties in regenerating complex injured tissues, when compared with mammalian species (63, 64), explaining why Zebrafish embryos and their stem cells have long been investigated to unravel cross-evolutionary regenerative mechanisms. The finding that hASC senescence can be reversed by a Zebrafish embryo extract shows that despite an evolutionary distance of about 450 million years human stem cells can sense ancestral microenvironmental cues and retrieve unexpected cell regulatory patterns. This consideration is also consonant with the observation that human cord-blood CD34+ cells are recruited into early vasculogenesis upon transplantation in pre-, but not post-gastrulation Zebrafish embryos (65).
Tissue aging likely involves the progressive senescence and potential exhaustion of tissue resident stem cells, leading to age-dependent decline in our ability to cope with tissue injury. Here, we show that the treatment of hASCs with early developmental stage factors from Zebrafish embryos may represent a useful method to counteract and reverse the transcriptional drop of a number of major anti-senescence conductors, retrieving relevant differentiating pathways. In future studies, considering the hurdle of using xenogenic material for preconditioning cells prior to transplantation in humans, we are planning to identify the active factors that may recapitulate the antisenescence action of ZF1, being potentially amenable for translation into clinical setting.
Federica Facchin and Silvia Canaider contributed equally to this article. The statistician Barbara Bordini is gratefully acknowledged for her contribution.
Abbreviations: hMSCs: human Mesenchymal Stem Cells; ZF1: early developmental stage Zebrafish embryo extract; hASCs, human Adipose-derived Stem Cells; NANOG: Nanog homeobox; POU5F1: POU domain class 5 homeobox 1, alias OCT4; MYC: v-myc avian myelocytomatosis viral oncogene homolog; BMI1: BMI1 proto-oncogene polycomb ring finger; TERT: telomerase reverse transcriptase; CDKN2A: cyclin-dependent kinase Inhibitor 2A, alias p16; hpf: hours post fertilization; LPA: Lysophosphatidic Acid; iPS: induced Pluripotent Stem; REAC: Radio Electric Asymmetric Conveyer; SA beta-gal: senescence-associated beta-galactosidase; h: hours; SD: standard deviation; p: passages;SOLV: solvent; HPRT1: hypoxanthine phosphoribosyl transferase 1; TBP: TATA box binding protein; GAPDH: glyceraldehyde 3-phosphate dehydrogenase; SEM: Standard error of the mean; alpha-MEM: alpha-Minimal Essential Medium; FBS: Fetal Bovine Serum; SDS-PAGE: Sodium Dodecyl Sulfate - Polyacrylamide Gel Electrophoresis; LC-MS/MS: Liquid Chromatography tandem-Mass Spectrometry; dH2O: deionized water