
Academic Editors: Simona Daniele and Rebecca Piccarducci
This is an open access article under the CC BY 4.0 license.
Background: Testicular germ cell tumors (TGCTs), a group of heterogeneous neoplasms, are the most frequent tumors of teenagers and young men, with the incidence rising worldwide. High cure rates can be achieved through cisplatin (CDDP)-based treatment, but approximately 10% of patients present refractory disease and virtually no treatment alternatives. Here, we explored new strategies to treat CDDP-resistant. Methods: In vitro TGCT CDDP-resistance model was established and differential mRNA expression profiles were evaluated using NanoString technology. Then, TGCT cell lines were treated with four potential drugs (PCNA-I1, ML323, T2AA, and MG-132) to overcome CDDP-resistance. Results: We found several differentially expressed genes related to DNA repair and cell cycle regulation on CDDP-resistant cell line (NTERA-2R) compared to parental cell line (NTERA-2P), and the proteasome inhibitor MG-132 demonstrated cytotoxic activity in all cell lines evaluated, even at a nanomolar range. MG-132 also enhanced cell lines’ sensitivity to CDDP, increasing apoptosis in both NTERA-2P and NTERA-2R. Conclusions: MG-132 emerges as a potential new drug to treat CDDP-resistant TGCT. Targeted therapy based on molecular mechanism insights may contribute to overcome acquired chemotherapy CDDP-resistance.
Testicular germ cell tumors (TGCTs) are a group of heterogeneous neoplasms resulting from a defective primordial germ cell development and are currently considered the most common tumors of teenagers and young men (15–40 years), representing 0.4% of new cases from all sites, with increasing incidence rates worldwide [1, 2, 3, 4].
TGCTs are classified into two main histological subtypes, including seminoma and non-seminoma germ cell tumors (NSGCT), representing 60% and 40% of the cases, respectively [5, 6]. NSGCTs are subdivided into several histologies, such as embryonal carcinoma, yolk sac tumors (YST), teratoma, choriocarcinoma, and mixed NSGCT [5, 6]. Thus, TGCTs are frequently heterogeneous tumors identified by their specific histology.
Platinum-based treatment is frequently used as first-line therapy for pediatric and adult TGCTs, and international guidelines have established a consensus for its application [7, 8]. Indeed, TGCTs are highly curable, exhibiting one of the highest sensitivity levels to platinum-derived compounds, and present an overall disease-free survival rate of approximately 80% for metastatic disease [9]. Even patients with advanced metastatic disease can achieve complete remission through systemic treatment and secondary resection of residual masses [10]. Its unique sensitivity is likely multi-factorial and associated with the germ cell origins of these tumors. Additional factors are known that may help explain this feature: TGCTs present a hypersensitive apoptotic response to DNA damage agents; show a decrease in the repair capacity of platinum-induced damage; only around 1% of TGCTs have TP53 mutations [11, 12]. Notably, however, about 10–20% of TGCT patients with advanced disease are refractory to platinum-based chemotherapy and have a less favorable prognosis with relapses, and treatment options for this group are extremely limited [13]. Studies have shown that late relapses occur more frequently in non-seminomas (3.2%) compared to seminomas (1.4%), in which the most often histological components are teratomas (60%) and YST (47%) [13, 14, 15]. Recent encouraging advances have been published in this area [16, 17, 18], but still, approximately 3–5% of all TGCT patients will eventually die of their disease [19, 20, 21].
Several reports have described molecular mechanisms related to cisplatin (CDDP)
resistance of TGCTs, including TP53 and MDM2 alterations [22],
induction of differentiation [23], global and specific DNA methylation
alterations [24], deregulation of the PDGFR
TGCT is a complex disease with various histological and clinical characteristics, so identifying specific molecular features critical for CDDP response will likely be necessary to effectively use this drug and/or find new treatment strategies [19].
Here, we established a TGCT model of resistance to CDDP, created after CDDP long-term exposure of parental NTERA-2 cell line, to identify mechanisms that are central for acquired CDDP-resistance in TGCTs. We performed an extensive phenotypic and molecular characterization of NTERA-2R and found that several genes related to DNA repair and cell cycle regulation are differentially expressed on resistant cells in response to CDDP. Moreover, the proteasome inhibitor MG-132 demonstrated cytotoxic activity in all TGCT cell lines evaluated, even at a low nanomolar range. MG-132 also enhanced cell lines’ sensitivity to CDDP in both, NTERA-2P and NTERA-2R, indicating that it could be a potential new strategy to overcome TGCTs treatment failure.
Two TGCT cell lines obtained from the European Collection of Authenticated Cell
Cultures (ECACC) were used. NTERA-2 clone D1 (ECACC Cat# 01071221,
RRID:CVCL_3407) is a cell line derived from a human testicular embryonal
carcinoma and was first described in 1984 [32]. 577MF (ECACC Cat# 06011802,
RRID:CVCL_2290) is a cell line derived from a human testicular teratocarcinoma
and was first described in 1980 [33]. Both were cultured following ECACC
recommended conditions and incubated in a humidified atmosphere with 5% CO
Cisplatin (CDDP) (PHR1624; Sigma-Aldrich) was prepared at 5 mM in 0.9% NaCl. PCNA-I1 (SML0730; Sigma-Aldrich), ML323 (SML1177; Sigma-Aldrich), T2AA (SML0794; Sigma-Aldrich) and MG-132 (M7449; Sigma-Aldrich) were prepared at 10 mM in DMSO (Sigma-Aldrich). All drugs were aliquoted and stored at –20 °C until use.
Exponentially growing cells were seeded at a density of 5
IC
The NTERA-2R IC
Evaluation of combination treatment was performed using CDDP IC
Cells were seeded in triplicate in a 12-well plate at a density of 5
To evaluate cell migration capacity, the monolayer wound-healing assay was performed. Briefly, a confluent monolayer of cells was seeded in a 6-well plate, and a “scratch” with a p1000 pipet tip was made through the cell layer. After washing several times with PBS, a medium containing 10% FBS was added to each well. Two fields of each wound were photographed in regular periods, from 0 to 64 hours. The wound areas were measured using TScratch software [35]. The experiments were performed in biological and technical triplicate.
Flow cytometry analysis of apoptosis and cell cycle were performed with FITC Annexin V Apoptosis Detection Kit I (BD Biosciences) and BD Cycletest Plus DNA Kit (BD Biosciences), respectively. Cells were seeded in T25 flasks and treated 24 hours later. After 72 hours, the culture supernatant and the cells were collected, washed twice with 1X PBS, and the specific protocols were followed as recommended by the manufacturer. Cell data was collected using BD Accuri Cytometer. Unstained and single-stained controls were used for color compensation, and at least 10,000 events were collected for each sample. The analysis was performed after two independent experiments, using FCS Express 7 software (De Novo Software, Pasadena, CA, USA).
Expression profile of NTERA-2R and NTERA-2P was performed using nCounter Vantage 3D DNA Damage and Repair panel (Nanostring, USA) as previously reported [36], to evaluate features related to CDDP-resistance. This panel includes 180 genes involved in major DNA damage repair pathways, including base excision repair, nucleotide excision repair, mismatch repair, TLS, and other repair processes (available at: http://nanostring.com).
Total RNA from NTERA-2P and NTERA-2R was isolated in biological triplicate, using TRIzol reagent, according to the manufacturer’s protocol. Probe pools, hybridization buffer, TagSet, and 100 ng total RNA (quantified by Qubit 2.0 Fluorometer) were hybridized for 21 hours at 67 °C, followed by purification and RNA/probe complexes immobilization in nCounter PrepStation (Nanostring, USA) and cartridge scanning in Digital Analyzer (Nanostring, USA), according to the manufacturer’s protocol.
The nSolver Analysis Software version 4.0 (NanoString Technologies, Seattle, WA,
USA) was used for quality control assessment, and further steps were carried out
in the R statistical environment, version 3.6.3. Gene expression levels and
sample distribution were evaluated with quantro package, version 1.18.0 [37].
Data normalization and differential expression were performed in the
NanoStringNorm package, version 1.2.1.1 [38]. Data were quantile normalized, and
log2 transformed. Differentially expressed genes were defined by the thresholds
of fold change
Venn diagram was created utilizing Venny 2.1 (RRID:SCR_016561, available at: https://bioinfogp.cnb.csic.es/tools/venny/). The STRING database was used to predict interaction networks from gene expression analysis. Clustering was performed using the K-means clustering method (RRID:SCR_005223, available at: https://string-db.org) [40]. Functional annotation analysis was performed using DAVID (RRID:SCR_001881, available at: https://david.ncifcrf.gov/home.jsp) [41, 42, 43].
Total proteins were extracted after 24 hours of treatment using RIPA buffer,
including 10% protease and phosphatase inhibitors (Sigma-Aldrich). After 15 min
on ice, samples were centrifuged at 13,000 g for 30 min at 4 °C, and the
supernatant was collected. Protein concentration was determined using the Bio-Rad
Protein assay, based on Bradford method (Bio-Rad) according to the manufacturer’s
instructions. Twenty micrograms of protein were denatured at 95 °C for 5
min in 4X Laemmli buffer (Thermo Fisher Scientific), separated on NUPAGE 10 or
15% bis–tris gels at 90 V and transferred to 0.2
Data are presented as means
We first performed a cell viability assay to determine the half-maximal
inhibitory concentration (IC
 Fig. 1.
Fig. 1.CDDP viability effect on NTERA-2 and 577MF TGCT cell
lines. (A) Representative images of the cell lines (Objective: 20x). (B) CDDP at
increasing concentrations was used to treat cells for 72 hours, and cell
viability was evaluated. IC
To better understand CDDP-resistance mechanisms, we chose the most sensitive
cell line in our previous analysis (NTERA-2) and established a CDDP-resistance
model (NTERA-2R). After eight months of CDDP treatment with incremental doses,
IC
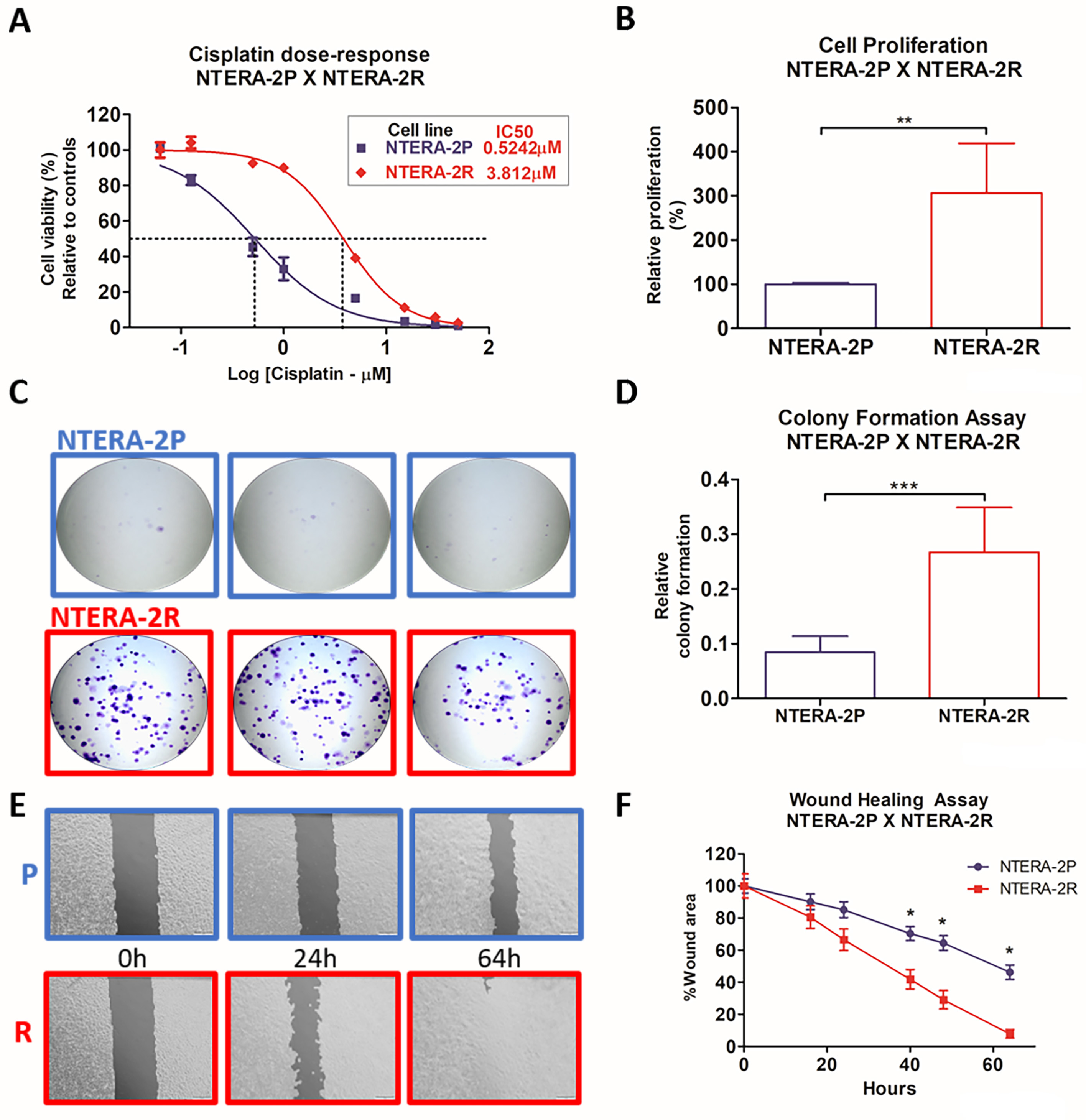 Fig. 2.
Fig. 2.Development and phenotypic characterization of TGCT
CDDP-resistance model. (A) Cell viability of NTERA-2P and NTERA-2R after 72
hours of CDDP treatment. IC
Levels of CDDP-induced apoptosis were assessed after 0.6
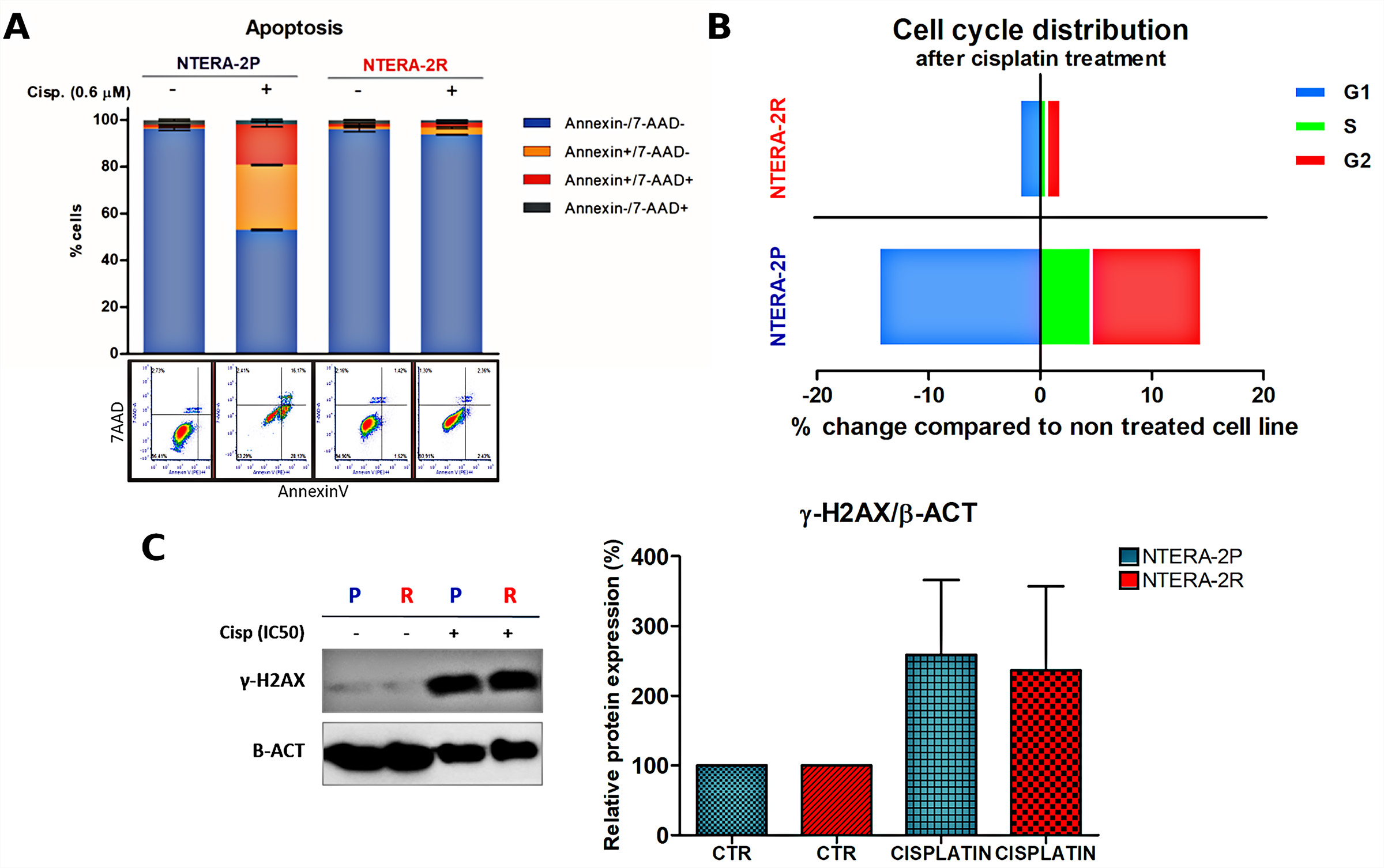 Fig. 3.
Fig. 3.Flow-cytometry analysis of CDDP effect on apoptosis and cell
cycle distribution. (A) apoptosis and (B) cell cycle distribution in NTERA-2P
and NTERA-2R, after 0.6
To elucidate the molecular changes associated with CDDP-resistance, we performed
an analysis of both NTERA-2P and NTERA-2R 72 hours after CDDP (IC
 Fig. 4.
Fig. 4.Differential expression analysis of CDDP-induced gene
expression. Nanostring DNA Damage and Repair panel revealed genes related to
CDDP-resistance in (A) NTERA-2P and (B) NTERA-2R. Only genes with fold change
A list with all genes differentially expressed (fold change
We used the STRING database to predict the interactions between the 24 genes. We
also used DAVID gene ontology (GO) to find the main regulated processes for these
genes. These analyses revealed two clusters mainly related to DNA repair
mechanisms and a cluster consisting of genes that regulate cell cycle, apoptosis,
and cellular signaling (Fig. 5A). DAVID Functional GO-Analysis identified nine
pathways significantly associated with the analyzed genes (p
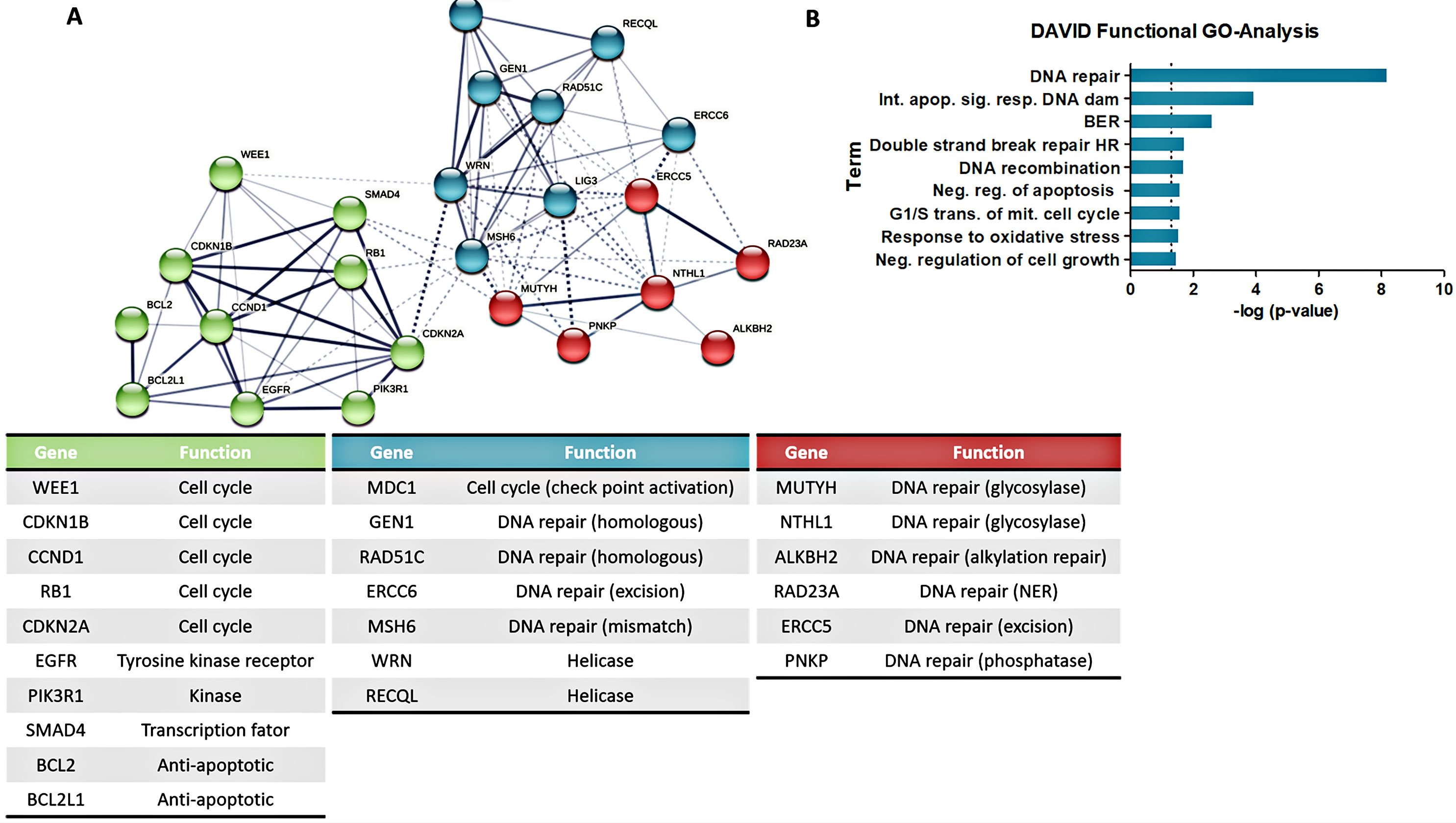 Fig. 5.
Fig. 5.Network and functional predictions of CDDP-induced gene
expression. Network and functional predictions indicate the main clusters and
pathways of the 24 genes exclusively altered in NTERA-2P and NTERA-2R cells after
CDDP treatment. (A) STRING interaction prediction considering the altered genes.
Genes primary function was identified, tabulated, and colors are according to
STRING clusters. (B) DAVID Functional GO-Analysis identified nine pathways
significantly associated with the analyzed genes (p
Considering the central role of cell cycle regulation and DNA repair mechanisms
in our CDDP-resistance model, we hypothesized whether a panel of drugs containing
three TLS inhibitors and one proteasome inhibitor, which was previously described
as targeting TLS, could be an alternative to overcome GCT resistance. We
performed a cell viability assay to determine the IC
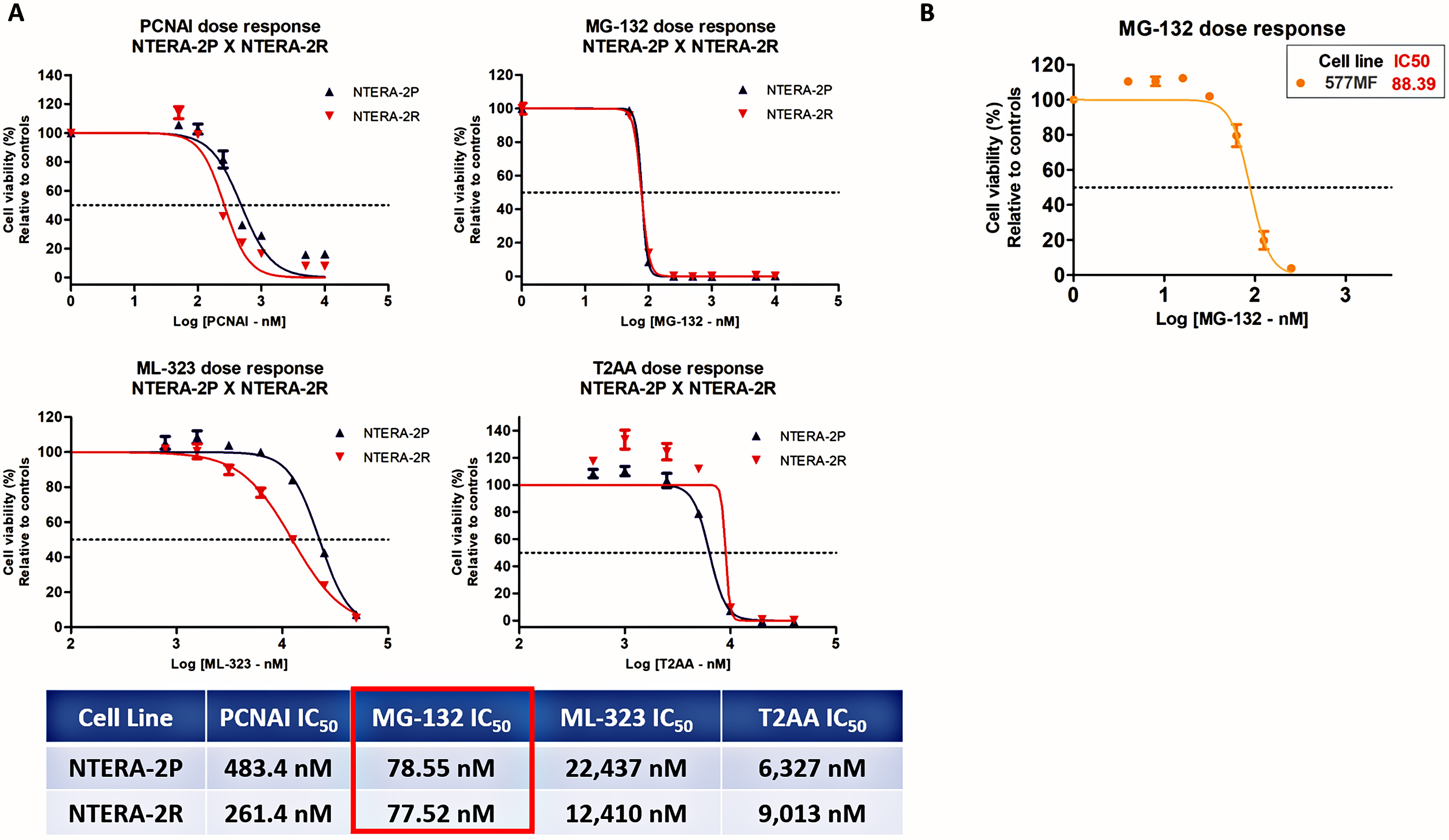 Fig. 6.
Fig. 6.Effect of a panel of drugs in TGCT cell lines viability. (A)
IC
In light of these data, we tested the potential of MG-132 and CDDP as a
combinatory treatment. We first assessed cell viability treating NTERA-2P and
NTERA-2R with a range of MG-132 concentrations combined with CDDP (IC
 Fig. 7.
Fig. 7.MG-132 and CDDP combination indicate great therapeutic potential
in both NTERA-2P and NTERA-2R. (A) Cell viability assay after MG-132 or CDDP
(IC
CDDP has a significant role in TGCTs treatment, yet a small subset of patients exhibit CDDP-resistance, constituting a critical clinical challenge [44]. Therefore, understanding the molecular mechanisms involved in CDDP-resistance of TGCTs and use this information to identify novel treatment alternatives for these patients is considered a major goal.
To address this objective, we first established and characterized an in
vitro model of TGCT CDDP-resistance (NTERA-2R). NTERA-2R cell line was
established using incremental CDDP doses (NTERA-2P: IC
CDDP-resistance mechanisms have been categorized, for organizational purposes,
as pre-target, on-target, and post-target, based on the following events after
the cell’s exposition to the drug [46]. We verified that NTERA-2P and NTERA-2R
expressed comparable amounts of the DNA damage sensor
On the other hand, on-target mechanisms have been extensively studied on TGCTs and connected to CDDP-resistance. These mechanisms are related to alterations that implicate DNA adducts formed upon CDDP binding, as DNA repair systems, or alternatively, the bypass of DNA adducts through a system known as translesion DNA synthesis (TLS) [47]. Here, we profiled the gene expression changes in both NTERA-2P and NTERA-2R using the NanoString DNA damage and repair panel. Among the five genes differentially expressed between NTERA-2P and NTERA-2R, we observed an increase in MGMT and XPC expression in NTERA-2R, which were already described to be involved in CDDP-resistance [48, 49, 50]. Moreover, MGMT expression levels also significantly correlated with a worst disease-free survival in TGCT cohort from the TCGA database, demonstrating it may be a promising biomarker of CDDP-resistance or even a therapeutic target for TGCT CDDP-resistance. Changes in the expression of POLD4 and POLE genes may indicate the involvement of mechanisms of TLS in our CDDP-resistance model. We also observed that the six genes more upregulated in NTERA-2R after CDDP treatment were also significantly upregulated in NTERA-2P CDDP treated. However, the most downregulated gene in NTERA-2R after CDDP treatment (ALKBH2) was exclusively observed in this cell line and may indicate a new mechanism of TGCT CDDP acquired resistance, as previously demonstrated in lung cancer cells [51]. In the analysis using STRING database and DAVID gene ontology we observed two clusters associated with DNA repair mechanisms and cell cycle, apoptosis, and cellular signaling. Besides, we found several strongly connected genes related to DNA repair and cell cycle regulation, which led us to hypothesize whether TLS could be related to TGCT CDDP-resistance. TLS has been associated with CDDP-resistance in other tumors with basically a dual role, suggesting that TLS inhibition may sensitize tumors to therapy as well as prevent the emergence of tumor chemoresistance [27, 52]. Besides, it has recently been acknowledged that the TLS is an important mechanism that presents a rationale to be further explored in TGCT CDDP-resistance [31].
TLS inhibitors may be an alternative to overcome CDDP-resistance, and the
targets can be diverse. The most obvious target though, is the replication
sliding clamp Proliferative Cell Nuclear Antigen (PCNA), which is considered the
primary regulator of TLS and one of the critical non-oncogenic mediators
supporting cancer development [53, 54]. In this study, we have chosen three
putative TLS inhibitors and one proteasome inhibitor, to evaluate their capacity
to inhibit cell proliferation of TGCT cells. (1) PCNA-I1 binds to PCNA and has
been shown to reduce the chromatin-associated PCNA in cells, and inhibits the
growth of many types of tumors [55]. (2) T2AA is an inhibitor of the PCNA/PIP-box
interaction [35] and can inhibit the growth of cancer cells through the induction
of early apoptosis [56]. (3) ML-323 is an effective inhibitor of the USP1-UAF1
deubiquitinase complex, responsible for deubiquitinating PCNA [57, 58]. ML-323 has
been shown to potentiate CDDP cytotoxicity in non-small cell lung cancer and
osteosarcoma cells and may be an alternative for overcoming CDDP-resistance [57].
(4) MG-132 is a peptide aldehyde, which effectively blocks the proteolytic
activity of the 26S proteasome complex and has been demonstrated to prevent TLS
in human cancer cells but not in normal cells [59]. However, the exact mechanism
by which MG-132 inhibits TLS remains to be elucidated. Here, we demonstrated that
the four TLS inhibitors’ present the capacity to reduce TGCT proliferation, even
of NTERA-2R cell line. MG-132 exhibited the highest cytotoxic potential, with
IC
We further explored the MG-132 potential by testing a combination treatment with CDDP to evaluate its capacity to improve CDDP-based therapy. We found that MG-132 enhanced CDDP activity in TGCT, triggering changes in cell cycle distribution and increased cell death through apoptosis, corroborating studies in ovarian carcinoma and osteosarcoma [60, 61].
This is the first pre-clinical study reporting the effectiveness of MG-132 in TGCT treatment, focusing on its potential to overcome CDDP-resistance alone or in combination with CDDP. It is worth mentioning that most of our results were obtained in the NTERA-2 cell line, which is a major limitation in our research. TGCTs are classified into two histological groups, including seminoma and NSGCTs, which includes embryonal carcinoma [5, 6]. It has been known that seminomas are highly sensitive to DNA-damaging agents, whereas non-seminomas including embryonal carcinoma and yolk sac tumor, often appear in the metastatic setting resistant to cisplatin, which reflects differences in cell biology [62, 63]. Thus, because NTERA-2 cell line is an embryonal carcinoma, the results reported here represent only this histological subtype. Another drawback of our study is that we did not develop other TGCT CDDP-resistant cell lines, which may limit our conclusions of MG-132 effectivity in TGCT CDDP resistance. In order to overcome this limitation, we evaluated the effects of MG-132 on the 577MF TGCT cell line, which was 5.5-fold more resistant to CDDP than NTERA-2P. Besides, 577MF has a different histology (teratocarcinoma), indicating MG-132 potential in histologies other than embryonal carcinoma. Even so, given the complexity and variability of TGCT, the role of MG-132 as a potential new drug to treat CDDP-resistant TGCT should be further evaluated in other cell lines to confirm our findings.
In future studies, we plan to explore in more detail whether and how these drugs may be involved in TGCT CDDP-resistance, assess its potential in TGCT cell lines from different histologies to broadly cover TGCT complexity, and use an in vivo TGCT model to evaluate their effectiveness and assess their toxicity profile. These may reveal the most appropriate approach to clinical applications and whether a specific biomarker will be necessary for this new therapy.
In this study, we demonstrated for the first time the potential of MG-132 used alone or in combination with CDDP to treat CDDP-resistant TGCTs. We developed a CDDP-resistance in vitro model and profiled the differentially expressed genes that may be related to the resistance phenotype. We showed the possibility of using new drugs to overcome TGCT CDDP-resistance and found that MG-132 displayed a potent cytotoxic activity against TGCT cells. Moreover, when used in combination with CDDP, MG-132 reduced cell viability and induced apoptosis and changes in cell cycle phases distribution. These results indicate that MG-132 is a potential treatment strategy to overcome TGCT treatment failure.
AvHL—Conceptualization, Formal analysis, Methodology, Validation, Writing – original draft. DOV—Conceptualization, Resources, Writing – review & editing. MTP—Conceptualization, Resources, Supervision, Writing – review & editing. RMR—Conceptualization, Resources, Writing – review & editing. LSdS—Formal analysis, Writing – review & editing. LdNBP—Methodology, Writing – review & editing. ERMC—Methodology, Validation, Writing – review & editing. INFG—Methodology, Validation, Writing – review & editing. LMdJ—Methodology, Writing – review & editing. MFSG—Methodology, Writing – review & editing. AOdR—Methodology, Writing – review & editing. TAT—Methodology, Writing – review & editing. ACL—Methodology, Writing – review & editing. LFL—Supervision, Writing – review & editing. All authors have read and agreed to the published version of the manuscript.
Not applicable.
The authors would like to thank André Lopes Carvalho and Matias Eliseo Melendez for gently providing TLS inhibitors.
This study was partially supported by the Public Ministry of Labor Campinas (Research, Prevention, and Education of Occupational Cancer) and Barretos Cancer Hospital. L.S.S. is the recipient of a grant from the Public Ministry of Labor Campinas (Research, Prevention and Education of Occupational Cancer). R.M.R. and L.F.L. are recipients of CNPq (National Council for Scientific and Technological Development) Productivity Grants. I.N.F.G. is recipient of a FAPESP (Sao Paulo Research Foundation) grant (2017/22305-9).
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.