
Academic Editors: Yingqun Wang, Haihua Feng and Graham Pawelec
Background: Pediatric brain tumors are the leading cause of cancer death in children and represent a variety of diseases and molecular subtypes. This study sought to evaluate a rapid immunohistochemistry testing panel to aid in therapy selection at the time of malignant tumor recurrence. Methods: With IRB approval and appropriate informed consent, we conducted a single-institution prospective clinical trial of selected kinase inhibitor therapy. A laboratory-developed immunohistochemical testing panel was performed on tumor tissue, and therapy with one of four small molecule inhibitors was recommended in combination with oral chemotherapy consisting of temozolomide and etoposide. Results: All 20 enrolled subjects were assigned to Everolimus (n = 4), Erlotinib (n = 6) or Dasatinib (n = 10); 90% (18/20) within the pre-specified 14-day feasibility time period. Only two subjects elected treatment on study, 8 received targeted treatment based on testing results either alone (n = 5) or in combination with chemotherapy (n = 3). Other subjects received chemotherapy alone (n = 7), surgery alone (n = 2) or no further therapy (n = 3). Immunohistochemical targets were associated with correlative genetic changes in 28% (5/18) of those evaluated. Conclusions: It was feasible to rapidly select targeted therapy in recurrent pediatric brain tumors, but not feasible to treat with a uniform combination treatment regimen.
Pediatric brain tumors are the leading cause of cancer death in children, with no standard effective therapy for patients who relapse. That being said, pediatric brain tumors are not a single uniform group, but are instead a heterogeneous group of rare neoplasms with many molecular drivers and subtypes, which have been defined in recent years [1]. The sheer number of new molecular diagnoses in pediatric brain tumors can make it challenging to design clinical trials, since many molecular subtypes are uncommon, and it may be challenging to enroll sufficient numbers of patients to evaluate efficacy within a specific histologic tumor type. At the same time, histology agnostic therapies have emerged such as PD-1 inhibitors and NTRK-inhibitors which have proven effective for a variety of tumor types [2, 3]. Many other tyrosine kinase inhibitors have also been developed and basket trials such as the Pediatric MATCH are underway to evaluate the tumor response of these drugs in wide range of tumors (ClinicalTrials.gov Identifier: NCT03155620). The median progression free survival for pediatric patients with recurrent CNS tumors enrolled on phase II studies of single-agent therapy is less than two months [4, 5, 6, 7, 8, 9].
Temozolomide and etoposide are an attractive combination as backbone chemotherapy because of their oral route of administration, and different mechanisms of action with preclinical data suggesting synergy between alkylating agents and topoisomerase inhibitors [10, 11, 12]. A phase I study established safe dosing of this combination even in heavily pre-treated patients, with hematologic dose-limiting toxicity [13]. In a prospective trial of this combination in 11 pediatric and young adult patients with high-grade brain tumors, five patients responded with tumor shrinkage, and an additional five patients achieved prolonged stable disease. Responding patients included diffuse midline glioma, CNS embryonal tumor, NOS, glioblastoma, and anaplastic astrocytoma [14].
In a review of pediatric patients with recurrent gliomas treated with oral etoposide and temozolomide using various dosing schedules, Korones et al. [15] reported responses in 7 of 11 patients, including a partial response in a patient with brainstem glioma and a complete response in a patient with glioblastoma. The rate of response in these preliminary combination studies appears to be much better than single-agent studies of either drug [8, 16, 17, 18].
The primary objective of this study was to determine the feasibility of utilizing data from rapid immunohistochemical studies performed on patient tumor tissue to inform treatment decisions in the setting of relapsed or refractory pediatric brain tumors. Secondary objectives were to estimate the objective response rate in patients who received biologically directed therapy in combination with chemotherapy, estimate event free and overall survival for pediatric patients who receive biologically directed therapy, and to further describe toxicity and tolerability of the combination regimens used in this study. Given prior experience with targeted next-generation sequencing [19, 20], correlative testing was performed to compare clinical sequencing to clinical immunohistochemical testing.
Potential protein biomarkers were initially screened using immunohistochemical stains to archived formalin-fixed paraffin-embedded tissue microarrays. Tissue microarrays included a variety of high grade primary pediatric brain tumors including initial diagnosis of medulloblastoma (n = 28), ependymoma (n = 17), high-grade glioma (n = 10), and CNS embryonal tumors, NOS (n = 6). In addition, 13 paired tumors obtained at the time of relapse included 2 medulloblastoma, 8 ependymoma, 1 high-grade glioma and 2 CNS embryonal tumors. Laboratory-developed immunohistochemical testing was performed for the following proteins: EGFR, HER2, CKIT, PDGFRA, pS6, pERK and scored using the methods detailed below. All patients included in these initial screening studies had appropriate consent and IRB approval was obtained for this analysis. Survival analysis was performed using data extracted from the electronic medical record system. The log-rank test was used to evaluate the significance of associations between survival, tumor type, age at diagnosis, extent of resection, and immunohistochemical staining pattern. Stepwise Cox regression was used to conduct multivariable analysis. Hazard of death associated with each biomarker was adjusted for tumor histology, age, and extent of resection.
This study was conducted as a prospective phase 1 clinical trial. FDA risk determination was requested regarding complex laboratory-developed testing, and the immunohistochemical panel was determined to pose a non-significant risk in the context of this study. The study was subsequently approved by the Seattle Children’s Institutional Review Board (IRB). Informed consent, and assent where applicable, was obtained from study participants or their legal guardians according to institutional standards, including IRB-approved process for phone consent for patients not residing in the region.
Eligibility for tumor testing included age less than 30 years, Karnofsky or
Lansky performance score
Treatment was recommended but not required for patients consented to study.
Treatment consisted of 28-day cycles of oral temozolomide 150 mg/m
All cases were reviewed by the study pathologist to confirm the diagnosis. The schema for assignment of treatment arm (see Fig. 1) was based on specificity of targeted agent with more specific agent selected if target was present. If more than one tumor specimen was available from different surgical procedures, the results of the most recent relapse specimen were prioritized. Additionally, if a sample was positive for more than one marker the drug priority was first Erlotinib, then Dasatinib, Everolimus, and last Sorafenib. Priority was based on drug specificity from most specific to less specific.
 Fig. 1.
Fig. 1.CONSORT diagram with study arm assignment.
Four-
Stains were interpreted as either positive or negative according to the
following algorithm. Each stain was first scored according to both staining
intensity (weak, moderate, to strong) and based on the percentage of tumor cells
staining positive (0 for no staining, 1+ for
The study was subsequently amended to offer optional correlative targeted tumor Next Generation Sequencing (NGS). Sequencing was performed using UW Oncoplex, a clinically validated method as previously reported [21]. This panel was designed to detect most classes of mutations, including single nucleotide variants, small insertions and deletions, gene amplifications, and selected gene-fusions. Sequencing libraries were prepared from DNA samples and hybridized to a custom set of complementary RNA (cRNA) biotinylated oligonucleotides targeting the exons of 262 cancer related genes and select intronic regions. NGS was performed using a HiSeq 2500 instrument system (Illumina, San Diego, CA, USA).
Response was evaluated by MRI imaging every two cycles. For subjects with
measurable disease, protocol-defined tumor measurements included the product of
the longest diameter and the next longest perpendicular measurement. Complete
response was defined as the disappearance of all abnormal signal, partial
response was defined as
The statistical design of the study defined two feasibility endpoints, namely (1) the feasibility of rapid testing, and (2) the feasibility of subjects receiving treatment on assigned study arm. Biologic selection by immunohistochemical testing was defined as feasible if at least 80% of study subjects received results of testing within two weeks of study enrollment. To meet this endpoint, tumor blocks were identified locally or acquired from another institution, tissue quality had to be adequate in amount and quality for staining and interpretation by study pathologist. Treatment on study was considered to be feasible if at least 50% of subjects started therapy with an assigned treatment arm. The treatment feasibility endpoint therefore depended on both the presence of at least one of the pre-specified protein targets as well as patient and/or treating physician decision-making.
In the archival tissue microarray studies eighty-nine percent of patients (54 of
61) evaluated had at least one positive immunohistochemical marker. The percent
of positive tumors was 16% for EGFR, 25% for HER2, 31% for KIT, 73% for
PDGFRA, 43% for pERK and 54% for pS6. Each marker was positive in a subset of
tumors of each histologic type, with the exception of HER2, which was negative in
all high-grade gliomas; and EGFR, which was negative in all ependymomas. In
addition to tumor histology, survival was associated with extent of resection and
expression of 3 of the 6 antigens evaluated: EGFR, PDGFRA and pS6 (log rank
p
Twenty subjects were enrolled on this phase 1 clinical trial. One additional subject was screened and found to be ineligible due to no documented disease progression following most recent medical therapy. Subject demographics and prior therapy are detailed in Table 1. Median age at the time of enrollment was 8 years (range 1–28 years), and 11 subjects (55%) were male. All patients had received prior multi-modality therapy. The number of prior surgeries was one in eight subjects, two in eight subjects, and three in four subjects. All 20 subjects received prior radiation therapy, including seven (35%) with prior craniospinal radiation and six subjects (30%) who had received two prior radiation therapy courses. Fifteen subjects (75%) received at least one prior chemotherapy regimen. The subjects without prior chemotherapy included diagnoses of ependymoma (n = 4) and chordoma (n = 1) who had received treatment with surgery and radiation.
| All subjects, results and treatment arm assignment | Immunohistochemical testing results | Correlative targeted Sequencing | |||||||||||||
| ID | Age (years) and gender | Tumor location and histology | Prior radiation | Prior chemo-therapy | Prior surgeries | Time to results (days) | Study Arm: Targeted therapy recommended | EGFR | HER2 | CD117 | PS6 | PDGFRA | ERK | Mutations | Copy Changes |
| 1 | 5 F | PF medulloblastoma | CSI | yes | 1 | 11 | D: dasatinib | – | – | – | + | + | + | sequencing not done | |
| 2 | 28 M | PF medulloblastoma | CSI | yes | 3 | 12* | D: dasatinib | – | – | + | + | + | + | sequencing not done | |
| 3 | 22 F | ST anaplastic astrocytoma | CSI | yes | 2 | 12* | D: dasatinib | – | – | + | + | + | + | H3F3A G35R, BCOR, TP53, ATRX | PDGFRA amp, MYCN amp, CDKN2A del |
| 4 | 14 M | ST anaplastic astrocytoma | focal | yes | 1 | 9 | D: dasatinib | – | – | – | + | + | + | H3F3A G35R, TP53, ATRX, PDGFRA, PTEN | CDKN2A del |
| 5 | 9 F | ST anaplastic pilocytic astroctyoma | focal | yes | 2 | 10 | D: dasatinib | – | – | – | + | + | + | MSH6 biallelic, POLE, hypermutated | |
| 6 | 4 F | PF ependymoma | focal, CSI | yes | 3 | 14* | B: everolimus | – | – | – | + | – | + | RAD51C | |
| 7 | 6 M | ST anaplastic astrocytoma | focal, focal | yes | 2 | 6 | C: erlotinib | + | – | – | + | – | + | NF1 | EGFR amp, MYCN amp, CDKN2A del, PTEN del |
| 8 | 8 M | ST glioblastoma | focal | yes | 1 | 17 | D: dasatinib | – | – | – | + | + | + | H3F3A K27M, PI3KCA, TP53 | NF1 loss |
| 9 | 7 M | PF ependymoma | focal, SRS | no | 1 | 19* | B: everolimus | – | – | – | + | – | + | negative | |
| 10 | 12 M | PF medulloblastoma | CSI | yes | 2 | 14* | D: dasatinib | – | – | – | + | + | + | TP53, PBRM1 | MYCN gain |
| 11 | 14 M | ST glioblastoma | focal, focal | yes | 2 | 12* | C: erlotinib | + | – | – | + | + | + | BCOR, KDM6A, MTOR, PDGFRA, PIK3CA, PIK3R1 | EGFR amp, CDKN2A loss |
| 12 | 8 M | PF ependymoma | focal | no | 1 | 7* | C: erlotinib | – | + | – | + | + | + | NOTCH2 | PTEN loss |
| 13 | 23 F | ST anaplastic astrocytoma | focal | yes | 3 | 6 | D: dasatinib | – | – | + | + | + | + | IDH1, ATRX, TP53 | |
| 14 | 19 F | Clivus chordoma | focal | no | 1 | 7* | C: erlotinib | + | – | + | + | + | + | TSC2 | TSC2 loss |
| 15 | 7 F | ST embryonal | CSI | yes | 2 | 7* | B: everolimus | – | – | – | + | – | + | MYCN gain | |
| 16 | 13 M | PF ependymoma | focal, focal | no | 3 | 22* | B: everolimus | – | – | – | + | – | + | negative | |
| 17 | 5 M | PF medulloblastoma | CSI | yes | 1 | 14* | D: dasatinib | – | – | + | + | + | + | FGFR3 amp, TACC3 amp, CDH1 loss | |
| 18 | 5 F | ST glioblastoma with embryonal features | focal | yes | 2 | 9* | C: erlotinib | –/+ | –/– | –/+ | + | –/+ | + | TP53 biallelic | MYCN amp, CDKN2A del |
| 19 | 7 F | PF ependymoma | focal | no | 2 | 12* | C: erlotinib | + | + | – | + | + | + | negative | |
| 20 | 1 M | PF atypical teratoid/rhabdoid tumor | focal, spinal | yes | 1 | 6 | D: dasatinib | – | – | – | + | + | + | SMARCB1 | |
| Key: M, Male; F, Female; PF, posterior fossa; ST, supratentorial; CSI,
craniospinal radiation; SRS, steriotactic radiosurgery; *includes time to obtain
tissue from other centers; amp: amplification; del: homozygous deletion; loss:
single copy loss; gain: copy gain of | |||||||||||||||
The rapid return of testing results was considered feasible, as results were returned within the 14-day study target goal in 90% of subjects (n = 18/20). Median time to return of results was 11.5 days (range 6–22 days). Only seven study subjects (35%) had tissue available at the primary study institution, and tissue was obtained from an outside institution for the remaining 13 subjects (65%). Testing was returned for the two subjects who did not meet the 14-day study target goal at 19 and 22 days. The delay to testing results was due to time to obtain tissue from outside institution in both cases. Targeted sequencing was performed for 18 subjects (90%), and oncogenic mutations or copy number changes were found in 15 of 18 tested (83%). Details of patient characteristics, prior treatment, and testing results are provided in Table 1. The highest levels of protein overexpression (3–4+) were often observed to be associated with gene amplifications or pathway mutations (Fig. 2). In six cases, 33% of cases sequenced, the immunohistochemical staining results identified cases with genetic alterations predicted to respond to targeted drug therapy.
 Fig. 2.
Fig. 2.Examples of high immune positivity associated with gene amplification, subject 3. (A) PDGFRA overexpression associated with gene amplification; subject 7 (B) EGFR overexpression associated with gene amplification; subject 14 (C) pS6 overexpression associated with TSC2 mutation.
No subjects were assigned to treatment arm A (Sorafenib). Four, six, and ten
subjects were assigned to treatment arms B (Everolimus), C (Erlotinib) and D
(Dasatinib), respectively. While all 20 subjects were assigned to a study
treatment arm (see Fig. 1), only two subjects (10%) elected treatment with
study-assigned therapy within the pre-specified four-week time-period (subjects 2
and 5). Of the two subjects who elected treatment on study, only one was able to
receive kinase inhibitor; the other subject experienced a delay in start of
targeted therapy due to insurance denial of targeted agent, and subsequently
experienced disease progression while receiving oral chemotherapy alone. Two
subjects received study-directed therapy off study, one who elected for other
treatment with re-irradiation prior to
initiation of study testing-directed
therapy (subject 7); the other decided not to travel for study therapy but
received treatment at home institution (subject 3). Both subjects remained on
therapy for 9 months until disease progression. Subject 3 experienced initial
response to combination study treatment which was sustained for
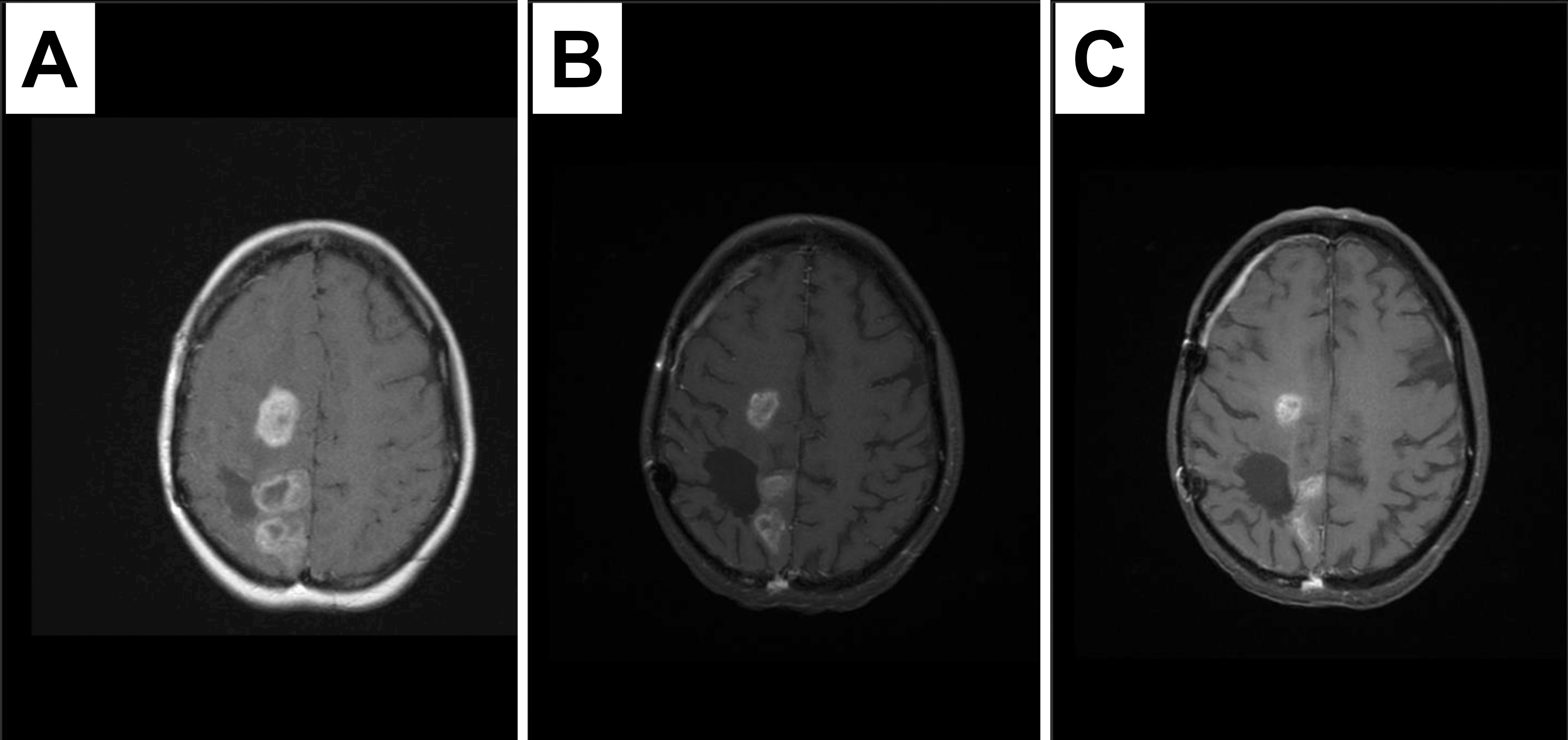 Fig. 3.
Fig. 3.Response to therapy containing dasatinib in a recurrent tumor with PDGFRA immunopositivity and amplification. MRI images at study enrollment (A), 3 months later demonstrating response (B) sustained at six months (C). Patient remained on therapy for 9 months until disease progression.
Five other subjects received targeted therapy alone based on study testing results, without recommended concurrent chemotherapy, one of whom received two sequential targeted therapies (subject 14). While a total of 8 subjects (40%) received targeted therapy based on study results, only one (5%) met the second feasibility endpoint according to study design. Details of subject therapy and follow-up are provided in Table 2.
| Treatment and follow-up | Treatment after study arm assignment | ||||||||||
| ID | Tumor location and histology | Study Arm: Targeted therapy recommended | Study treatment, on study | Study treatment, not on study | Targeted Only | Other chemo only | Surgery Only | No treatment | Description of Targeted Therapy | Status | Follow-up time (months) |
| 1 | PF medulloblastoma | D: dasatinib | + | none, combination chemotherapy | DOD | 11 | |||||
| 2 | PF medulloblastoma | D: dasatinib | +* | none*, temozolomide/etoposide only | DOD | 3 | |||||
| 3 | ST anaplastic astrocytoma | D: dasatinib | + | dasatinib with temozolomide/etoposide | DOD | 9 | |||||
| 4 | ST anaplastic astrocytoma | D: dasatinib | + | none, no tumor-directed therapy | DOD | 0 | |||||
| 5 | ST anaplastic pilocytic astroctyoma | D: dasatinib | + | dasatinib with temozolomide/etoposide | DOD | 2 | |||||
| 6 | PF ependymoma | B: everolimus | + | everolimus | DOD | 7 | |||||
| 7 | ST anaplastic astrocytoma | C: erlotinib | + | erlotinib with temozolomide/etoposide | DOD | 10 | |||||
| 8 | ST glioblastoma | D: dasatinib | + | none, no tumor-directed therapy | DOD | 3 | |||||
| 9 | PF ependymoma | B: everolimus | + | none, re-resection only | NED | 32 | |||||
| 10 | PF medulloblastoma | D: dasatinib | + | vorinostat | SMN | 13 | |||||
| 11 | ST glioblastoma | C: erlotinib | + | none, etoposide | DOD | 1 | |||||
| 12 | PF ependymoma | C: erlotinib | + | everolimus | DOD | 10 | |||||
| 13 | ST anaplastic astrocytoma | D: dasatinib | + | none, no tumor-directed therapy** | NED** | 15 | |||||
| 14 | Clivus chordoma | C: erlotinib | + | imatinib, everolimus | DOC | 6 | |||||
| 15 | ST embryonal | B: everolimus | + | everolimus | AWD | 21 | |||||
| 16 | PF ependymoma | B: everolimus | + | everolimus | AWD | 25 | |||||
| 17 | PF medulloblastoma | D: dasatinib | + | none, combination chemotherapy | AWD | 25 | |||||
| 18 | ST glioblastoma with embryonal features | C: erlotinib | + | none, other study therapy | DOD | 8 | |||||
| 19 | PF ependymoma | C: erlotinib | + | none, re-resection only | NED^ | 0^ | |||||
| 20 | PF atypical teratoid/rhabdoid tumor | D: dasatinib | + | none, combination chemotherapy | DOD | 4 | |||||
| Key: PF, posterior fossa; ST, supratentorial; DOD, death from disease; DOC, death from other causes; AWD, Alive with disease; NED, no evidence of disease;*prescribed dasatinib, but insurance denied and patient never received, **progression later attributed to pseudoprogression, ^subject lost to follow-up after initial return of results. | |||||||||||
The remaining subjects who did not receive targeted treatment received either chemotherapy alone (n = 7), surgery alone (n = 2) or no further tumor-directed therapy (n = 3). One subject who did not plan initial post re-resection therapy was subsequently lost to follow-up after return of study results. At the time of study completion, 12 subjects had died of disease, one died of secondary malignancy (acute myelogenous leukemia), one died of infectious complications of surgery, and three were alive with disease. Only two patients were in follow-up with no evidence of disease, one of whom was later deemed to have probable pseudoprogression.
We found that it was feasible to select kinase inhibitor therapy based on a limited panel of oncogenic protein targets utilizing a rapid laboratory-developed immunohistochemical panel. Although the majority of subjects on study did not have tissue available at the local study institution, it was feasible to obtain tissue suitable for rapid immunohistochemical evaluation in all patients on study, 90% within the target window of 14 days, which allowed for the incorporation of testing results when selecting therapy in the setting of recurrent pediatric malignancy. Correlative targeted sequencing was also feasible to obtain in all subjects in whom it was attempted as optional testing. NGS in general takes significantly longer than immunostaining, so we were not able to use the sequencing results in assigning patients to a treatment arm, however such technologies are improving rapidly and other pediatric brain tumor trials currently in progress are using tumor sequencing to assign treatment (ClincalTrials.gov Identifiers: NCT02724579, NCT03581292).
Despite availability of testing results in a timely manner, the study did not meet the second feasibility endpoint regarding compliance with study-prescribed treatment. We found that the study “one size fits all” approach to backbone low-dose chemotherapy was not appealing to many subjects, for a variety of reasons including the desire and ability to receive care closer to home. Most subjects selected either chemotherapy alone or targeted therapy alone rather than the study combination recommended. Many local physicians were willing to prescribe off-label targeted therapies. This practice supports patients and families remaining near home rather than traveling to a tertiary treatment center. Interestingly, and perhaps because nearly all standard medical therapy in recurrent pediatric brain tumors consists of off-label medication use, in only one case was insurance denial a treatment-limiting factor. It is also of note that only one of the subjects enrolled on this trial selected treatment on a different clinical trial, likely due to lack of clinical trial options in this heavily pre-treated group.
It is a challenge to evaluate the efficacy of any precision medicine approach, and this study was not designed or powered to be able to address an efficacy question. While the study of efficacy of a single targeted therapy for rare mutations in a specific subtype is in theory feasible, it would be wholly impractical to study each rare mutation or disease separately (e.g., Everolimus in TSC-mutated chordoma). Other clinical trials are also evaluating this histology agnostic personalized approach [22, 23]. This trial does demonstrate the high prevalence of potential targets for therapy in children with recurrent brain tumors [19]. Further study of the efficacy of a selected or precision medicine approach to therapy is warranted, whether through prospective novel clinical trial design or larger collaborative population studies.
This study protocol was developed during a time of controversy over the role of the FDA in regulation of laboratory-developed testing, just prior to the release of initial FDA draft guidance on laboratory developed testing (LDT). At the instruction of the local Institutional Review Board, the study team sought FDA determination regarding the risk of the LDT used in this study, and ultimately received a non-significant risk determination from the FDA allowing the trial to proceed as designed. As laboratory testing becomes more complex, the role of the FDA versus the Center for Medicaid and Medicare (CMS) Clinical Laboratory Improvement Amendments (CLIA) remains a controversy affecting the implementation of clinical trials of precision medicine.
While we observed a few anecdotal early signals of targeted therapy efficacy in children on this trial, the majority ultimately died from progression of recurrent therapy-resistant brain tumors, which remain the leading cause of cancer death in children. Much of brain tumor biology has been described in the past decade, and it currently remains unclear whether we need better targets for therapy and/or better therapies for known targets [24, 25, 26, 27, 28, 29, 30]. Novel mechanisms to target tumor-related proteins such as antibody therapy, vaccine therapy, and cellular immunotherapy are under evaluation against some of the same targets identified in this study, and going forward we plan to support our institution in clinical trials using CAR-T cells and other novel approaches (ClinicalTrials.gov Identifiers: NCT03638167, NCT03500991) [31]. These therapies may hold greater potential compared to the previously available small molecules without good CNS penetration. As our understanding of pediatric brain tumor biology and molecular subtypes changes, so will clinically relevant laboratory-developed testing. In addition, the decreasing cost and turnaround time of multiplexed genomic testing has gained acceptance and has been incorporated into clinical care.
In this phase 1 clinical trial we found rapid testing by immunohistochemistry was feasible for screening tumors and selecting kinase inhibitor therapy for patients. In addition, the highest levels of protein overexpression were often observed to be associated with gene amplifications or pathway mutations identified by sequencing. Despite availability of testing results in a timely manner, the study did not meet the second feasibility aim regarding compliance with study-prescribed treatment for a variety of reasons detailed in our manuscript. This work supports the further clinical development of targeted therapeutics that may inhibit growth of tumors based on these proteins, such as immunotherapy approaches.
All authors contributed significantly to this project. Author SESL and KS designed the research study. Authors SESL, BLC, KS, and CML all performed parts of this research. SESL and BLC analyzed the data. SESL and BLC wrote the manuscript. All authors contributed to editorial changes, read, and approved the final manuscript.
This study was approved by the Seattle Children’s Institutional Review Board (IRB). Informed consent, and assent where applicable, was obtained from study participants or their legal guardians.
The authors wish to thank Russ Geyer for his mentorship and Erin Rudzinski for her advice and support. We also wish to thank the Seattle Children’s Research Laboratory Services for their work on this study.
We thank the Seattle Children’s Hospital Cancer Research Pilot Funds and Clinical Research Scholars Program for funding this project. This research received no funding external to our institution.
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.