1. Introduction
Radiation facilities and radioactive materials are used extensively in the
military, industrial, medical and scientific fields, greatly increasing the
possibility of large-scale, uncontrolled exposure to radiation [1, 2]. As a
constantly renewing organ with rapidly proliferating and maturing cells, the skin
is sensitive to radiation [1, 2]. Ionizing radiation promotes the production of
reactive nitrogen and reactive oxygen species (RNA/ROS) due to the radiolysis of
water and direct ionization of target molecules; this increased production leads
to oxidative damage and skin injuries [3, 4]. Approximately 95% of cancer
patients treated with radiation develop some form of radiation dermatitis,
including erythema, dry desquamation, and moist desquamation [5, 6].
Radiation-induced skin damage has a negative impact on the effectiveness of
radiation therapy and the quality of life of patients [7]. Despite significant
improvements in radiation technology, radiation-induced skin toxicity remains a
problem [5, 6, 7, 8].
Peroxisome proliferator-activated receptors (PPARs) are ligand-inducible
transcriptional factors that belong to the hormone nuclear receptor superfamily.
Three members of the PPAR family (PPAR, PPAR/ and
PPAR) with a high degree of sequence homology have distinct
physiological roles, ligand specificity, and tissue distribution [9, 10].
PPAR is a vital regulator of fatty acid oxidation in a wide variety of
tissues [11, 12]. Fibrates are synthetic PPAR ligands, and they serve as
first-line drugs for reducing serum triglyceride levels [13, 14]. When activated,
nuclear-localized PPAR heterodimerizes with the retinoid X receptor and
binds to PPAR-responsive elements (PPREs), which consequently stimulate the
transcription of an extensive array of target genes associated with lipid
metabolism, cell differentiation, inflammation and many other biological
processes [15, 16]. PPAR agonists have been shown to confer protection
against various tissue injuries in a variety of radiation-induced injury models,
including radiation-induced brain injury and heart injury [17, 18]. In addition,
previous research has confirmed that PPAR agonists would ameliorate the
proinflammatory responses seen in the microglia following in vitro
radiation [19].
Fenofibrate, a specific ligand for PPAR, has long been used to treat
hypercholesterolemia, hypertriglyceridemia, diabetes and cardiovascular
diseases [14, 20]. Fenofibrate reduces low-density lipoprotein (LDL), very
low-density lipoprotein (VLDL), and triglyceride levels and increases
high-density lipoprotein (HDL) levels [14, 20]. PPAR also has
antioxidant and anti-inflammatory properties [13]. Fenofibrate confers
cytoprotective effects against myocardial ischemia-reperfusion (I/R) injury in
rats by suppressing cell apoptosis and attenuating age-related renal injury by
activating AMPK and SIRT1 signaling pathways [20, 21].
We have recently reported the beneficial effect of fenofibrate against
radiation-induced skin injury in animal models and human patients [22]. However,
its underlying mechanisms remain unknown. In this study, we demonstrated that
fenofibrate-induced PPAR activation conferred protection against
ionizing radiation to the skin. We identified fatty acid binding protein 4
(FABP4) as a key effector for fenofibrate-mediated protection against
radiation-induced ROS production and lipid accumulation. These results suggest
that fenofibrate protects against radiation-induced skin damage through FABP4.
2. Materials and Methods
2.1 Reagents
Dimethylsulfoxide (DMSO) and
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) were
purchased from Solarbio (Beijing, China).
Fenofibrate and the FABP4 inhibitor BMS309403
were obtained from Sigma-Aldrich (St. Louis, MO,
USA). 4’-6-diamidino-2-phenylindole (DAPI) and
Hoechst stains were purchased from Beyotime Biotech (Nantong, China). A
SmartFlare uptake control probe (positive control) and FABP4 mRNA-specific
SmartFlare probe were obtained from Millipore (Billerica, MA, USA). BODIPY
fluorophore 493/503 for lipid droplets was obtained from Molecular Probes
(Eugene, OR, USA). Adenoviruses (Ad-NC and Ad-FABP4) were obtained from HanBio
(Shanghai, China).
2.2 Animal Studies
Protocols for experiments involving animals were approved by the Animal
Experimentation Ethics Committee at Soochow University (Suzhou, China). Male
Sprague-Dawley (SD) rats (4 weeks old) and male C57 mice (4 weeks old) were
purchased from the Shanghai SLAC Laboratory Animal Co., Ltd. (Shanghai, China).
For irradiation, the rats were anesthetized with an intraperitoneal injection of
ketamine (75 mg/kg) and xylazine (10 mg/kg), and the hair on the rat buttocks was
shaved using a razor. A 3-cm-thick piece of lead was used to shield the rats and
localize the radiation field (3 cm 4 cm). A single dose of 45 Gy
irradiation [23, 24, 25] was administered to the hindlimb region of the SD rats
at a dose rate of 750 cGy/min using a 6-MeV electron beam accelerator (Clinac
2100EX, Varian Medical Systems, Inc., CA, USA). This dose was selected because it
can significantly induce skin injury [23, 24, 25, 26]. For the treatment, the rats were
then randomly assigned to receive treatments by subcutaneous injection of DMSO,
fenofibrate, or adenovirus [26].
2.3 RNA Extraction and Real-Time PCR Analysis
Total RNA was extracted from cells and tissues with Trizol reagent (Invitrogen,
Carlsbad, CA, USA). PPAR and FABP4 mRNA levels were
quantified by quantitative real-time PCR as reported previously [27]. The primers
used are listed in Supplementary Table 1.
2.4 Human Skin Samples
Human skin samples were obtained from a victim of an iridium radiation accident
as reported previously [28]. The skin samples were obtained 160 days after
irradiation from the right limb, which was exposed to iridium-192 (Ir)
metal chain (with an activity of 966.4 GBq or 26.1 Ci). Normal skin tissues were
obtained when performing skin grafting from the dorsal myocutaneous flap.
Informed consent for sample collection was obtained from the patient.
2.5 Immunohistochemistry (IHC)
Skin tissues from mouse, rat and monkey were obtained as reported previously
[24]. Mouse skin tissues were irradiated with 35 Gy electron beam. Rat skin
tissues were irradiated with 45 Gy electron beam. The skin tissues of monkeys
were treated with 0 or 20 Gy irradiation. Skin tissues were fixed in 10%
neutral-buffered formalin and embedded in paraffin. Three-micrometer paraffin
sections were deparaffinized and heat treated with citrate buffer (pH 6.0) for 7
min following an epitope retrieval protocol. Three-micrometer paraffin sections
were incubated with a rabbit anti-PPAR antibody (Abcam, Cambridge, MA,
USA, #ab 8934) at 4 °C overnight, followed by incubation with an
anti-rabbit biotinylated secondary antibody (Beyotime, Nantong, China),
diaminobenzidine substrate detection, washing, hematoxylin staining, dehydration,
and mounting.
2.6 Malondialdehyde (MDA) Concentration Measurement
Tissue MDA levels were determined using thiobarbituric acid (TBA) assays as
reported previously [24]. MDA levels were normalized to those of the control
group.
2.7 ROS Generation Assay
ROS levels were determined using the ROS-sensitive dye 2,7-dichlorofluorescein
diacetate (DCF-DA) (Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
The cells were washed with PBS and incubated with DCF-DA (10 M) for 30
min. Skin tissues were trypsinized into single cell suspension according to the
manufacturer’s instructions. The level of DCF fluorescence, which reflects the
ROS concentration, was observed with a fluorescence microscope. DCF fluorescence
levels in skin cells and tissues were quantified at 488 nm using a 96-well plate
reader.
2.8 Cell Culture and Irradiation
Human keratinocyte HaCaT cells, human fibroblast WS1 cells [24, 25, 26] and
primary skin fibroblasts were maintained in Dulbecco’s modified Eagle’s medium
(DMEM). All culture media was supplemented with 10% FBS (Gibco, Grand Island,
NY, USA). Cells were grown at 37 °C in 5% CO incubators. The
cells were exposed to different dosages of ionizing radiation using an X-ray
linear accelerator (Rad Source, Suwanee, GA, USA) and a fixed dose rate of 1.15
Gy/min.
2.9 Cell Viability Assay
Cells were incubated with DMSO or fenofibrate. Cell viability was measured using
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assays.
The cells were incubated for 4 h with 200 g/mL MTT (Sigma, St Louis, MO,
USA). The reagent was dissolved in DMSO (Solon, OH, USA). The absorbance values
were measured at 490 nm using a 96-well plate reader. The experiments were
performed in triplicate.
2.10 Immunostaining
Cells were fixed in 4% paraformaldehyde, washed with PBS, and permeabilized
with 1% Triton X-100 in PBS. The cells were then blocked with blocking buffer
(PBS, 1% Triton X-100, and 5% BSA) and incubated at 4 °C with a
PPAR (Abcam, #ab 8934) or H2AX antibody (Abcam; #ab 81299)
overnight. Next, a rhodamine-conjugated goat anti-rabbit antibody (1:100) was
added for 30 min at room temperature. The nuclei were counterstained with DAPI.
2.11 Luciferase Reporters and Luciferase Assay
The luciferase reporter with four PPREs in luciferase promoter was a
kind gift from Dr. Zengpeng Li (Third Institute of Oceanography, State Oceanic
Administration, Xiamen, China). The plasmid was verified by sequencing. Cells
were transfected with the constructed vectors using Fugene HD transfection
reagent (Promega, Madison, WI, USA). For each transfection, 50 ng pRL-TK
(Promega) was used to enhance the transfection efficiency. Measurement of
luciferase activity using the dual luciferase reporter assay system (Promega).
Promoter activity was expressed as the ratio of firefly luciferase activity to
Renilla luciferase activity.
2.12 Western Blotting Analysis
Detailed descriptions are given as previously described [25]. Briefly, the
membranes were blotted with antibodies against PPAR (Abcam, Cambridge,
MA, USA, #ab227074), GAPDH (Abcam, #ab181602), and FABP4 (Abcam, #ab 92501).
2.13 Measurement of Cell Apoptosis
Cells were pretreated with DMSO or fenofibrate and then exposed to irradiation.
Apoptosis was measured using a 7-AAD/Annexin-V double staining
apoptosis kit (BD Biosciences, Franklin Lakes, NJ, USA) and
flow cytometry (BD Biosciences, CA, USA). The Annexin-V+/7-AAD- cells indicated
early apoptosis, and the Annexin-V+/7-AAD+ cells indicated late apoptosis. The
percentages of both types of cells were counted.
2.14 Electromobility Shift Assay (EMSA)
WS1 cell nuclear protein was extracted using a nuclear protein isolation kit
(Beyotime). The sequences for the double-stranded oligonucleotide probes
(Supplementary Table 2) were synthesized and labeled with biotin by
Shanghai Sangon Biotech Co. Ltd. (Shanghai, China). EMSAs were performed
according to the LightShift EMSA Kit instructions (Pierce, Rockford, IL, USA).
2.15 Statistical Analysis
The data are expressed as the mean SEM of at least three independent
experiments. The results were evaluated via one-way ANOVA to determine
statistical significance. The statistical analyses were performed using Prism 8
(GraphPad software, San Diego, CA). The differences were considered significant
at p 0.05.
3. Results
3.1 Ionizing Radiation Decreases Cutaneous PPAR
Expression
We firs0074 analyzed the response of PPAR to ionizing radiation in
multiple animal models. Rats were irradiated with a 45 Gy electron beam as
reported previously [26, 27]. The real-time PCR analysis results showed that
PPAR mRNA levels in the irradiated skin tissues were 26.62%
of those in the nonirradiated skin tissues. This result is consistent with our
RNA-Seq data (GEO database accession number GSE86252) [28]. Next, we attempted to
confirm the expression of PPAR in skin tissue after irradiation by
immunohistochemistry in different animal models. The results showed that the
expression of PPAR in the skin tissues of mice, rats, and monkeys after
irradiation was significantly lower than that of the nonirradiated control group
(Fig. 1B). Moreover, in the irradiated epidermis of a human patient, the
expression of PPAR was decreased, with pronounced distribution from the
nucleus to the cytosol (Fig. 1C), indicating PPAR inactivation in
irradiated skin cells. In addition, ionizing radiation downregulated
PPAR protein levels in a dose-dependent manner in human skin fibroblast
WS1 and human keratinocyte HaCaT cells (Fig. 1D).
 Fig. 1.
Fig. 1.
Ionizing radiation decreases the expression of
PPARexpression in skin tissues. (A) PPAR
mRNA levels in irradiated and nonirradiated skin tissues of rats (n = 5). PPAR mRNA expression was measured by real-time PCR. The data
are shown as the mean SEM. (B) The expression of PPAR in
irradiated and nonirradiated skin tissues of mouse, rat and monkey. Skin tissues
were collected three days after indicated radiation doses. PPAR
expression was measured by IHC as described in the Materials and Methods section.
(C) The expression of PPAR in nonirradiated and irradiated human skin
tissues. (D) Western blotting analyses of PPAR expression at different
doses of radiation in WS1 and HaCaT cells. (E) Quantitative analysis of Western
blotting assay. Data are depicted as the mean SD from three independent
experiments. * p 0.05; ** p 0.01, compared with the
control group.
3.2 Fenofibrate-Mediated PPAR Activation Protects Skin
Cells against Radiation
Because fenofibrate is a synthetic fibrate ligand of PPAR, we next
explored its effect on PPAR activation and its influence on the
radiosensitivity of cultured skin cells. HaCaT cells were exposed to fenofibrate
and then subjected to immunofluorescence for PPAR detection. The
results indicated that fenofibrate induces the translocation of PPAR
into the nucleus (Fig. 2A). Moreover, the activity of the PPRE harboring
luciferase reporter was significantly increased after fenofibrate addition; this
result confirmed PPAR activation by fenofibrate (Fig. 2B).
 Fig. 2.
Fig. 2.
Fenofibrate-meditated PPAR activation protects skin
cells against radiation. (A) HaCaT cells were treated with 25 and 50 M
fenofibrate, and immunofluorescence was performed to investigate PPAR
translocation. (B) The effect of fenofibrate on PPAR activity was
measured with a PPRE luciferase reporter. Luciferase activity was assayed 24 h
after transfection. The firefly luciferase activity of each sample was
normalized to the Renilla luciferase activity. The final luciferase
activity was normalized to that of the control group. (C) HaCaT and (D) WS1 cells
were pretreated with fenofibrate and subjected to 0 or 20 Gy irradiation. One
hour later, the cellular ROS levels of each group of cells were determined using
a DCF-DA probe. Cellular fluorescence was observed using a fluorescence
microscope. ROS levels were quantified with a microplate reader. (E) HaCaT cells
were pretreated with DMSO or fenofibrate and then irradiated. Mitochondrial
membrane potential was evaluated using JC-1 staining. (F) HaCaT cells were
pretreated with 25 and 50 M fenofibrate. Then, the cells were mock
irradiated or irradiated with 20-Gy X-rays. Cell apoptosis rates were detected
with Annexin-V/7-AAD staining at (F) 48 h and (G) 72 h after irradiation. The
data are shown as the mean SEM of three independent experiments. (H)
HaCaT cells were treated with DMSO or fenofibrate. Representative
photomicrographs of BODIPY fluorophore 493/503 staining for lipid droplets. The
cells were observed with a confocal microscope (Olympus, Tokyo, Japan). (I)
Quantification of the ratio of JC-1 aggregate to JC-1 monomer and ATP contents.
(J) Quantification of BODIPY fluorophore 493/503 staining for lipid droplets.
Data are depicted as the mean SD from three independent experiments.
*p 0.05; ** p 0.01, compared with the control group.
Because ionizing radiation elicits cutaneous free radical
reactions [3, 4], we examined whether PPAR activation confers protection
against radiation-induced oxidative damage. Fenofibrate
concentrations of up to 50 M did not significantly affect viability in
HaCaT and WS1 cells (Supplementary Fig. 1). We first measured
fenofibrate effects on cellular ROS elimination in human HaCaT keratinocytes, WS1
fibroblasts and primary human fibroblasts. HaCaT cells pretreated with 25 or 50
M Fenofibrate significantly reduced radiation-induced ROS levels (Fig. 1C). Similar results were obtained in WS1 cells and human primary fibroblasts; in
these cells, 50 M fenofibrate exhibited the strongest antioxidative
activity (Fig. 2D and Supplementary Fig. 2).
Mitochondrial functional failure, involving mitochondrial membrane potential
changes is considered to be one of the most important factors leading to cell
death [29, 30]. Nonirradiated HaCaT cells were stained with JC-1 to show red
fluorescence, while a large number of cells switched to green fluorescence after
irradiation. These results indicate a decrease in mitochondrial membrane
potential. HaCaT cells treated with fenofibrate showed less fluorescence from red
to green, suggesting that fenofibrate can maintain mitochondrial membrane
potential after ionizing radiation (Fig. 2E). These results demonstrated that
fenofibrate protects mitochondria from ionizing radiation.
We next explored whether fenofibrate was associated with decreased apoptosis in
skin cells. As shown in Fig. 2E,F, fenofibrate did not affect apoptosis in HaCaT
cells that were not exposed to irradiation. In comparison, treatment with both 25
and 50 M fenofibrate significantly decreased apoptosis in HaCaT cells that
were exposed to 20 Gy irradiation (Fig. 2G). These results demonstrated that
fenofibrate-mediated PPAR activation reduces apoptotic cell death
caused by irradiation in skin cells.
Because epidermal lipids and free fatty acids play important roles in cell
growth, differentiation and permeability barrier function [31, 32], we
investigated whether fenofibrate-mediated PPAR activation modulated
lipid accumulation in human keratinocytes. The results revealed that
PPAR activation by fenofibrate increased cytoplasmic lipid accumulation
in HaCaT cells (Fig. 2H).
3.3 Fenofibrate Ameliorates Radiation-Induced Skin Injury in Rat
Model
Next, we sought to investigate whether fenofibrate could mitigate the
progression of radiation-induced skin injury in animal models. A
radiation-induced rat skin injury model (45 Gy electron beam irradiation)
[25, 26] was used to evaluate the role of clinically approved fenofibrate in
oxidative damage. After exposure to 45 Gy of irradiation, rat skin was injected
subcutaneously with DMSO or fenofibrate. To test whether fenofibrate affects
radiation-induced lipid peroxidation, we measured ROS and MDA concentrations in
skin tissues three days after 45 Gy of irradiation. As shown in Fig. 3A,B, both
cellular ROS and MDA levels were significantly lower in fenofibrate-injected
tissues than in DMSO-injected tissues. This result indicated that fenofibrate
attenuated radiation-induced ROS and consequent lipid peroxidation.
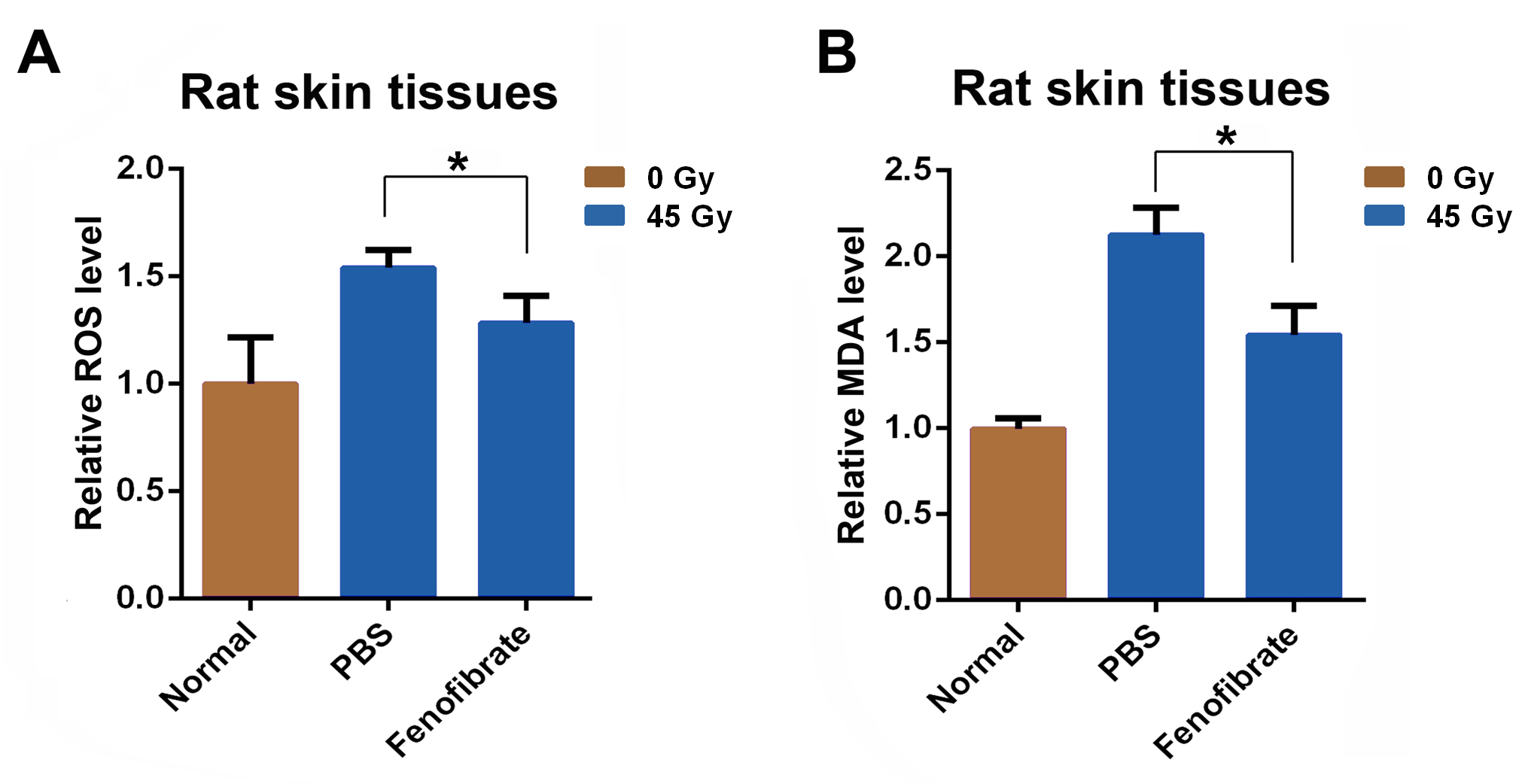 Fig. 3.
Fig. 3.
Fenofibrate ameliorates radiation-induced skin injury in a rat
model. Rat gluteal skin was unexposed or irradiated with a single dose from a
45-Gy electron beam. The rats were then randomly assigned to receive one of the
following treatments (n = 4): (1) subcutaneous 110 L DMSO injection (in
890 L PBS); (2) subcutaneous 400 g fenofibrate injection (in 110
L DMSO and 890 L PBS). (A) Relative ROS levels in the rat skin.
Three days after irradiation, skin ROS levels were determined as described in the
Materials and Methods section. (B) MDA concentration levels in rat skin from
different groups at three days after irradiation. p 0.05, compared
with the control group.
3.4 Fenofibrate Stimulates FABP4 Expression in Skin Cells
Our results showed that fenofibrate-mediated PPAR activation promoted
lipid accumulation in skin cells. This finding indicates a potential relationship
between skin cells and lipid metabolism. Free fatty acids, which are relatively
insoluble and potentially toxic, can be transported to other cells by
noncatalytic binding proteins [33]. FABPs belong to a family of intracellular
proteins and exhibit a high affinity for non-covalent binding to long-chain fatty
acids [34]. We, therefore, hypothesized that FABPs may be involved in the
radioprotective role of fenofibrate. Among the 12 identified members of the human
FABP family, three putative binding sites for PPAR (PPRE) in the
proximal promoter of FABP4 (Fig. 4A) were predicted by bioinformatics
analysis. This result suggested transcriptional regulation by PPAR.
FABP4 is an intracellular lipid-binding protein responsible for fatty acid
transportation [35] and we have recently shown that FABP4-mediated the
radioprotection of adipocytes [26]. We next performed EMSAs to investigate the
binding of potential transcriptional factors. EMSAs revealed that
oligonucleotides representing the predicted PPAR binding sites all
formed a specific complex when incubated with WS1 nuclear extracts (Fig. 4B).
Western blotting analyses showed that fenofibrate increased FABP4 protein levels
in both HaCaT and WS1 cells (Fig. 4C). Using real-time PCR analyses, we found
that fenofibrate increased FABP4 mRNA levels in a dose-dependent manner
(Fig. 4D). A FABP4 mRNA-based fluorescent probe, but not a SmartFlare uptake
control probe, confirmed that fenofibrate upregulated FABP4 transcripts
specifically in WS1 and HaCaT cells (Fig. 4E and Supplementary Fig. 3).
Taken together, these results clearly indicated that FABP4 is positively
regulated by the PPAR agonist fenofibrate in skin cells.
 Fig. 4.
Fig. 4.
Fenofibrate activates FABP4 expression in skin cells. (A)
Bioinformatics analysis predicted three putative binding sites in the proximal
promoter of FABP4. (B) EMSA using nuclear proteins from WS1 cells and
oligonucleotides carrying the indicated probes. Lanes 1, 3 and 5 contain the
probes without nuclear extracts. Lanes 2, 4 and 6 contain the oligonucleotide
probes 1, 2 and 3, respectively. (C) HaCaT and WS1 cells were treated with 10–50
M fenofibrate. FABP4 expression was measured by Western blotting analyses.
(D) WS1 cells were treated with 10–50 M fenofibrate, and FABP4
mRNA was quantified by real-time PCR. (E) Quantitative analysis of Western
blotting assay. (F) Quantification of FABP4-specific SmartFlare probe fluorophore
microscope (G) HaCaT and WS1 cells were treated with 50 M fenofibrate for
24 h, and FABP4 mRNA was detected with a FABP4-specific SmartFlare probe
(Millipore, Billerica, MA, USA). Fluorescent signals reflecting the
FABP4 mRNA levels were observed using a confocal microscope. p 0.05 and ** p 0.01, compared with the control group.
3.5 FABP4 Protects Skin Cells from Radiation-Induced Damage
Next, we sought to investigate whether increased FABP4 expression could modulate
radiation-induced damage in skin cells. Skin cells were pre-infected with a
control adenovirus (Ad-NC) or FABP4 overexpression adenovirus (Ad-FABP4) and
subjected to X-ray irradiation (Fig. 5A,B). The results showed that FABP4
overexpression reduced radiation-induced ROS levels (Fig. 5C). Moreover, FABP4
overexpression increased cellular lipid accumulation in HaCaT cells, which mimics
the effect of fenofibrate (Fig. 5D). Immunofluorescence assays for H2AX
foci showed that fewer foci were present in irradiated WS1 cells with FABP4
overexpression than in control cells (Fig. 5E). These data suggested that FABP4
facilitated the repair of radiation-induced DNA damage.
 Fig. 5.
Fig. 5.
FABP4 confers radioprotection to skin cells. WS1 cells were
infected with the indicated adenoviruses. (A) FABP4 expression was measured by
Western blotting analyses. (B) FABP4 expression was measured by Western blotting.
(C) WS1 cells were infected with the indicated adenovirus, followed by 0 or 20 Gy
irradiation. Cellular ROS levels for each group of cells were determined 1 h
after radiation using a DCF-DA probe and quantified with a microplate reader. (D)
Quantification of BODIPY fluorophore 493/503 staining for lipid droplets. (E)
Quantification of Western blotting assay. (F) Quantification of nuclear
H2AX foci fluorescent signals. (G) The effect of FABP4 overexpression
on lipid accumulation in HaCaT cells. Representative photomicrographs of BODIPY
fluorophore 493/503 staining for lipid droplets. (H) WS1 cells were infected with
Ad-NC or Ad-FABP4, and the dynamic repair process of DNA double-strand breaks
(DSBs) was measured by detecting nuclear H2AX foci after X-ray
irradiation. p 0.05 and ** p 0.01, compared with the
control group.
3.6 FABP4 Mediates the Radioprotective Role of Fenofibrate
To investigate whether FABP4 mediated the radioprotective role of fenofibrate,
FABP4 inhibitor BMS309403 [36] was used. The results showed that
the addition of BMS309403 exacerbated radiation-induced ROS in human skin
fibroblasts. Moreover, the ROS-eliminating activity of fenofibrate was abrogated
by BMS309403 (Fig. 6A). These results indicated that FABP4 was involved in
antioxidant response and that FABP4 mediated the ROS eliminating the role of
fenofibrate. Moreover, combined treatment with fenofibrate and BMS309403
abrogated lipid accumulation activity of fenofibrate in keratinocytes, which
suggested that FABP4 mediated fenofibrate-induced lipid accumulation (Fig. 6B). Taken together, these above results indicated that FABP4 was likely
to mediate the radioprotective role of fenofibrate.
 Fig. 6.
Fig. 6.
FABP4 mediates the radioprotective role of fenofibrate. (A) WS1
cells were infected with the indicated adenovirus or treated with fenofibrate
and/or BMS309403, followed by 0 or 20 Gy irradiation. Cellular ROS levels for
each group of cells were determined 1 h after radiation using a DCF-DA probe and
quantified with a microplate reader. (B) The FABP4 inhibitor BMS309403 abrogated
fenofibrate-induced lipid accumulation in HaCaT cells. Representative
photomicrographs of BODIPY fluorophore 493/503 staining for lipid droplets. (C)
Quantification of BODIPY fluorophore 493/503 staining for lipid droplets.
p 0.05, compared with the control group.
3.7 FABP4 Protects Skin from Radiation-Induced Damage In Vivo
Next, we investigated whether FABP4 overexpression could reduce
radiation-induced skin damage in vivo. The buttock region of rats was
irradiated with a 45 Gy electron beam to model the irradiation-induced skin
injury in rats. Irradiation at 45 Gy significantly increased skin ROS levels at
three days after treatment, as shown in Fig. 7A, ROS levels were significantly
lower in tissues infected with Ad-FABP4 than in the control tissues. Moreover,
FABP4 overexpression also reduced radiation-induced MDA levels (Fig. 7B). These
results indicate that FABP4 overexpression attenuates lipid peroxidation
resulting from radiation-induced oxidation.
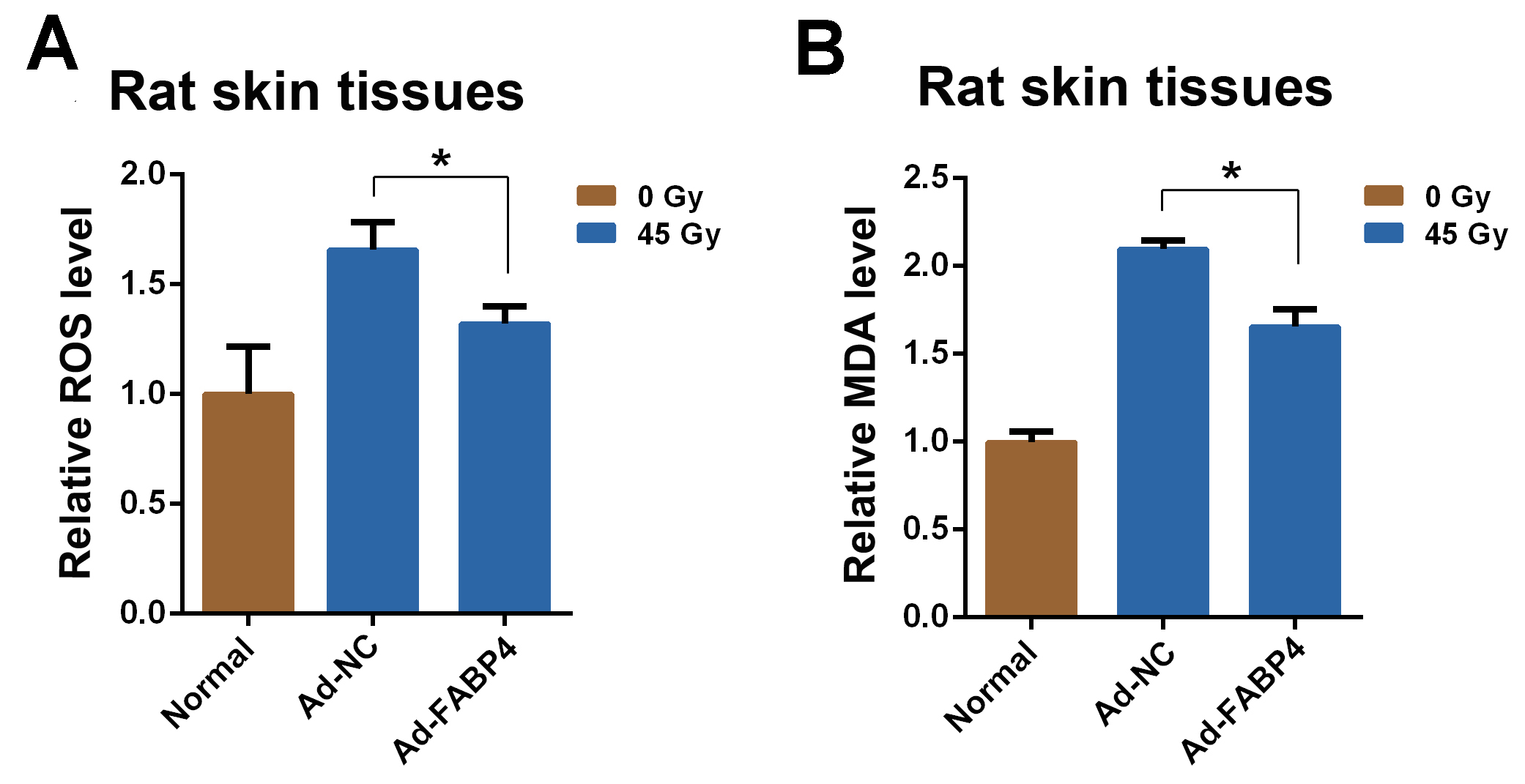 Fig. 7.
Fig. 7.
FABP4 attenuates radiation-induced skin injury in a rat model. Rat gluteal skin was unexposed or irradiated with a single dose from a 45-Gy
electron beam, followed by subcutaneous injection of Ad-NC (5 10
genomic copies of Ad-NC in a 200-L volume) or Ad-FABP4 (5
10 genomic copies of Ad-FABP4 in a 200-L volume) (n = 4). (A)
Relative ROS levels in rat skin. Three days after irradiation, skin ROS levels
were determined as described in the Materials and Methods section. (B) MDA
concentration levels in rat skin from different groups at three days after
irradiation. p 0.05, compared with the control group.
4. Discussion
Radiation-induced skin damage remains a serious problem following exposure to
ionizing radiation, including nuclear accidents, terrorist attacks, and radiation
therapy. However, there are currently only limited effective treatments to
prevent or mitigate radiation-induced skin damage [5, 6, 7]. Our previous report
indicates the involvement of the PPAR pathway in the response of skin tissues to
ionizing radiation [26]. The three different PPAR isotypes display distinct
physiological and pharmacological functions depending on their target genes and
tissue distribution [37, 38]. Although PPAR as a target for radiation is
well established in radiation research, especially in normal tissue injuries such
as heart, skin, and brain injuries, PPAR agonists have been shown to
confer tissue injury protection in a variety of radiation-induced injury models
[17, 18]. Previous research has established that persistent alteration of cardiac
metabolism due to impaired PPAR activity contributes to the heart
pathology after radiation [18]. We also have previously reported the beneficial
effect of fenofibrate against radiation-induced skin injury in animal models and
human patients [22], but its underlying mechanism remains elusive. In this study,
we found that fenofibrate-mediated PPAR activation reduced
radiation-induced ROS and apoptosis. Comparatively, equivalent amounts of the
PPAR agonist rosiglitazone [39] did not protect against
radiation-induced cutaneous damage in our study (data not shown), indicating a
PPAR-specific effect or that these specific rosiglitazone doses are
ineffective for this disease. Compared with that of PPAR, the function
of PPAR has been reported to be more restricted to fatty acid uptake
and -oxidation [10, 11, 12, 13, 14]. In addition, the antioxidant and
anti-inflammatory roles of PPAR activators have also been reported in
specific types of cells [21, 22]. For example, the PPAR agonist WY14643
improves homeostasis and the skin barrier function [40]. Fenofibrate has been
shown to reduce LPS-induced ROS through GCH1 in human umbilical vein endothelial
cells (HUVECs) [41]. We also have previously shown that GCH1 overexpression
reduces radiation-induced ROS by inhibiting NOS uncoupling in skin cells [25]. On
the other hand, research has confirmed that expression of heme oxygenase-1 (HO-1)
in human vascular cells is regulated by peroxisome proliferator-activated
receptors [42]. Our previous reports have provided further evidence that
increased HO-1 expression due to ionizing radiation suppressed ROS production and
reduced radiation-induced skin injury [26, 43]. In this study, it was found that
PPAR agonist can regulate target protein the expression of FABP4, and
it can regulate the expression of FABP4 through regulating lipid antioxidants to
reduce ROS production. However, the specific mechanism remains to be further
explored. Taken together, this study expands the beneficial application of
fenofibrate in treating human diseases.
PPAR, together with RXR, binds to a specific PPRE DNA sequence element
with a consensus sequence that consists of a direct repeat of the hexameric
sequence AGG(A/T) CA separated by one less well-conserved spacer nucleotide [40].
In this study, we identified FABP4 as a direct target of PPAR
activation in skin cells. This finding expands the list of
PPAR-regulated targets. Of all the FABPs, FABP4 possesses a unique high
affinity for both saturated and unsaturated fatty acids; this function has been
well characterized in cellular metabolism homeostasis [34, 35]. In addition, FABP4
has also been shown to promote cell growth and metastasis in multiple
malignancies, partially through supplying fatty acids and energy [44, 45]. We have
previously shown that FABP4 facilitates cell migration and the repair of
radiation-induced DNA breaks [26]. During wound healing, the skin often requires
more energy from the body’s energy stores to build new cells and restore the
barrier function [46]. PPAR activates FABP4, which can facilitate
cellular free fatty acid uptake, deliver essential fatty acids and provide an
energy supply for damaged cells. In addition, the skin needs lipids for rapid
cornification and the barrier function of the stratum corneum, which is present
as a lipid double layer in a lipid matrix [30, 31, 47]. Therefore, increased levels
of FABPs likely provide essential fatty acids for normal metabolism and skin
barrier function. Herein, we confirmed that fenofibrate/FABP4 increased lipid
accumulation in human keratinocytes. Another PPAR agonist, WY-14643,
has been shown to increase cellular lipids in keratinocytes in vitro and
in vivo, which is consistent with our finding [48]. Several skin
diseases, such as psoriasis and atopic dermatitis, are associated with reduced
skin lipids [49, 50]. Therefore, these findings may have significance not only for
radiation-induced skin injury but may represent one mechanism in cutaneous
diseases. Moreover, we also found that FABP4 mediated the ROS scavenging role of
fenofibrate. Thus, PPAR/FABP4 constitutes a novel strategy to
ameliorate radiation-induced skin injury. However, the molecular mechanism of
FABP4 in eliminating ROS warrants further investigation.
5. Conclusions
In summary, we found that PPAR agonist fenofibrate confers
radioprotection by stimulating FABP4 in skin cells (Fig. 8). These findings
provide a potential strategy for treating radiation-induced skin injury.
 Fig. 8.
Fig. 8.
Schematic representation of PPAR agonist fenofibrate
confers radioprotection by stimulating FABP4 in skin cells. PPAR
agonist fenofibrate induced PPAR expression in the irradiated skin
cells, which results in the proximal promoter of fatty acid binding protein 4
(FABP4) harbored three binding sites recruitment of PPAR and stimulated
the transcription of FABP4 in skin cells. FABP4 activation significantly
decreased radiation-induced oxidative damage in vivo.
Abbreviations
PPAR, peroxisome proliferator-activated receptor ; ROS,
reactive oxygen species; FABP4, fatty acid binding protein 4.
Author Contributions
SZ and JZ conceived and designed the study. CS, BS, and DY carried out the
molecular biology studies. WS and FG performed the animal experiments. SZ and YJ
drafted the manuscript and the figures. YJ, TY and KF performed statistical
analyses. All authors read and approved the final manuscript.
Ethics Approval and Consent to Participate
Ethical approval was obtained from the Ethics Committee of Soochow University
(approval number: 2016-0101).
Acknowledgment
Thanks to all the peer reviewers for their opinions and suggestions.
Funding
This work is supported by the National Natural Science Foundation of China
(82073477, 31770909, 81773226 and 32071238), Military Logistics Research Program,
Innovation Project of Chengdu (2021-YF05-01603-SN), the Young Talent Project of
China National Nuclear Corporation and Central Government Funds of Guiding Local
Scientific and Technological Development for Sichuan Province (No. 2021ZYD0073).
Conflict of Interest
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
