
Academic Editor: Graham Pawelec
Background: In this study, the entire mitochondrial genome (mitogenome) of Aleuroclava psidii (Singh, 1931) (Hemiptera: Aleyrodidae) was sequenced. The species A. psidii is currently classified in the subfamily Aleyrodinae. This mitogenome is the first representative from the genus Aleuroclava. Methods: Next-generation sequencing was used to obtain the molecular data. We conducted phylogenetic analyses with 18 existing mitogenomes of whiteflies and three outgroups of psyllids, under the Maximum likelihood and Bayesian inference criteria. Results: The arrangement of genes differed between the mitogenome of A. psidii and the putative ancestral insect mitogenome, and also differed from the mitogenomes of other whiteflies. Mitochondrial gene rearrangements involved the transpositions of trnQ, trnY, and the protein-coding gene nad1. Most hemipteran mitogenomes have the same mitochondrial gene order as that inferred to be ancestral for insects. However, there are an increased number of gene rearrangements in the mitogenomes of whiteflies. Phylogenetic reconstructions supported Aleurodicinae and Aleyrodinae as being monophyletic. Conclusions: Comparison of the gene order of mitogenomes revealed a clade-specific evolutionary trend in whiteflies. This study demonstrates the potential of using structural rearrangements to resolve major phylogenetic relationships within Aleyrodidae.
Mitochondrial genome (mitogenome) sequences and structure have been widely used to investigate the phylogenetic relationships of insects [1, 2, 3]. The insect mitogenome is a small and generally circular, double-stranded molecule with a genome size ranging from 15,000 bp to 18,000 bp [1]. Typically, the insect mitogenome contains 37 genes encoding 13 protein-coding genes, 22 transfer RNA genes, and two ribosomal RNA genes. A large non-coding region is often present between the small ribosomal RNA gene and the trnI gene. This region usually has a higher A+T content and contains elements for transcription and/or replication of the mitogenome [1, 4, 5, 6], and is thus also called the AT-rich region or control region. The entire mitogenome of insects is compact and the intergenic spacer sequences are usually very short.
The mitochondrial gene order in insects generally remains stable among most groups. The gene arrangement of the fruit fly Drosophila yakuba Burla, 1954 is considered to be ancestral for insects [7, 8]. The large majority of insect species therefore have an identical arrangement of mitochondrial genes as D. yakuba [8]. Nevertheless, discrete lineages have been discovered that exhibit many more mitochondrial gene rearrangements than other groups, such as the parasitic wasps in Hymenoptera [9, 10], some thrips in Thysanoptera [7, 11], and the lice in Phthiraptera [12, 13]. Most groups within Hemiptera have a mitogenome organization that is identical to the proposed ancestor, with just several exceptional gene rearrangements reported in the suborders Auchenorrhyncha [14, 15] and Heteroptera [16, 17, 18, 19]. In contrast to other hemipterans, however, all of the published mitogenomes of whiteflies show an extraordinary number of rearrangements. Moreover, the gene arrangements vary between different groups of whitefly species. Despite the fact that some aspects of whitefly mitogenomes are unusual, there is still no description of the characteristics of these genomes in a phylogenetic framework.
Whiteflies are a group of insects classified in the family Aleyrodidae of the suborder Sternorrhyncha within Hemiptera. The family Aleyrodidae includes many serious agricultural pests, such as Bemisia tabaci (Gennadius, 1889). This is an economically important invasive species that causes considerable damage to agricultural crops. In addition to directly feeding on plants, some whitefly species can also transmit plant viruses, with one study suggesting that 114 virus species are transmitted by whiteflies [20].
Mitochondria are key eukaryotic organelles involved in metabolism, apoptosis, disease and aging in animals [21, 22, 23, 24, 25]. While mitochondrial oxidative phosphorylation is the major source of ATP for the cell [21], each of these processes may be associated with the biological characteristics of some insects. Sequencing and characterization of complete mitogenomes provides a key to address the evolution of mitochondria. To date (January, 2022), the mitogenome sequences of only 18 whitefly species have been reported in GenBank. The limited data has so far limited efforts to reconstruct a comprehensive phylogeny of this insect group and thus better understand the evolution of mitogenomes.
In the present study, we provide an additional mitogenome sequence from the family Aleyrodidae, namely Aleuroclava psidii (Singh, 1931). This is the first complete mitogenome sequence of the genus Aleuroclava. The two main objectives of this study were: (1) to investigate the phylogenetic relationships within Aleyrodidae using mitogenome sequences, and (2) to assess the phylogenetic utility of mitochondrial gene rearrangements for this group.
Specimens of A. psidii (about 50 adults) were collected from photinia
(Photinia
Genomic DNA was extracted from a pool of 40 specimens using the TIANamp Micro DNA Kit (Tiangen Biotech Co., ltd., Beijing, China). The DNA quality and concentration were examined with a nucleic acid protein analyzer (Quawell Technology Inc., San Jose, CA, USA).
Genomic DNA was sonicated to 300 bp using a Covaris S220 focused-ultrasonicator
(Covaris Inc., Woburn, MA, USA), according to the Illumina protocol. Library
generation for Illumina Hiseq sequencing was carried out using the Illumina
TruSeqTM DNA Sample Prep Kit (Illumina, San Diego, CA, USA). The resulting
fragment library was sequenced on an Illumina HiSeq2500 platform, with a strategy
of 150 paired-end sequencing. Data quality control was performed using the NGS QC
Toolkit v2.3.3 (New Delhi, India) [28], under the default settings. Adapters,
ploy-N, and low-quality reads were removed from raw data. The high-quality reads
(avg. Q20
Two assemblers were used for mitogenome assembly, namely GetOrganelle v1.7.5.2 (Kunming, China) [29] and Geneious R11 (Auckland, New Zealand) [30]. For assembly with GetOrganelle [29], we used the GetOrganelle animal database (-F animal_mt) to identify, filter, and assemble target-associated reads. For assembly using Geneious [30], we applied the “map to reference strategy”, with the pre-sequenced mitochondrial cox1 gene sequence as the reference. The parameter settings were selected as described by Yang et al. [31].
The newly sequenced mitogenome of A. psidii as well as mitogenome sequences obtained from GenBank were annotated in the MITOS webserver [32] (http://mitos2.bioinf.uni-leipzig.de/index.py, Leipzig, Germany) to obtain uniform annotations. The RefSeq 63 Metazoa and the Invertebrate genetic code were selected as job settings. Mitogenome structure images for all whitefly species were generated using OGDRAW (Potsdam-Golm, Germany) [33]. Gene boundaries for protein-coding genes were refined by alignment with closely related whiteflies (Table 1). Sequences of protein-coding genes were separately aligned using TranslatorX (London, UK) [34]. A detailed description of parameter settings for TranslatorX [34] is available in the section on Sequence alignment. The secondary structures of 22 tRNA genes were predicted by MITOS [32]. The gene boundaries of two rRNA genes were refined by alignment against published sequences. The rRNA sequences were aligned separately using MAFFT version 7 (Osaka, Japan) [35]. The secondary structures of rRNA genes were inferred by reference to D. yakuba [36]. The annotated mitogenome sequence was submitted to GenBank under the accession number OM362911.
| Superfamily | Family | Subfamily | Species | GenBank accession |
| Aleyrodoidea | Aleyrodidae | Aleurodicinae | Aleurodicus dugesii | AY521251 |
| Aleyrodoidea | Aleyrodidae | Aleurodicinae | Aleurodicus dispersus | KR063274 |
| Aleyrodoidea | Aleyrodidae | Aleyrodinae | Aleuroclava psidii | OM362911 |
| Aleyrodoidea | Aleyrodidae | Aleyrodinae | Aleurocanthus spiniferus | KJ437166 |
| Aleyrodoidea | Aleyrodidae | Aleyrodinae | Aleurocanthus camelliae | KU761949 |
| Aleyrodoidea | Aleyrodidae | Aleyrodinae | Aleurochiton aceris | AY572538 |
| Aleyrodoidea | Aleyrodidae | Aleyrodinae | aff. Aleurochiton sp. | MH999477 |
| Aleyrodoidea | Aleyrodidae | Aleyrodinae | Aleyrodes shizuokensis | MT880225 |
| Aleyrodoidea | Aleyrodidae | N/A | Aleyrodidae sp. | MK645228 |
| Aleyrodoidea | Aleyrodidae | Aleyrodinae | Bemisia tabaci | AY521259 |
| Aleyrodoidea | Aleyrodidae | Aleyrodinae | Bemisia afer | KF734668 |
| Aleyrodoidea | Aleyrodidae | Aleyrodinae | Bemisia emiliae | KX714967 |
| Aleyrodoidea | Aleyrodidae | Aleyrodinae | Bemisia sp. | KX714968 |
| Aleyrodoidea | Aleyrodidae | Aleyrodinae | Neomaskellia andropogonis | AY572539 |
| Aleyrodoidea | Aleyrodidae | Aleyrodinae | Pealius mori | LR877884 |
| Aleyrodoidea | Aleyrodidae | Aleyrodinae | Pealius machili | MT015588 |
| Aleyrodoidea | Aleyrodidae | Aleyrodinae | Singhiella simplex | LR877885 |
| Aleyrodoidea | Aleyrodidae | Aleyrodinae | Tetraleurodes acaciae | AY521262 |
| Aleyrodoidea | Aleyrodidae | Aleyrodinae | Trialeurodes vaporariorum | AY521265 |
| Psylloidea | Liviidae | N/A | Diaphorina citri | KU647697 |
| Psylloidea | Psyllidae | Acizziinae | Acizzia uncatoides | MG989217 |
| Psylloidea | Psyllidae | Psyllinae | Arytainilla spartiophila | MG989220 |
| Note: Bold indicates the species sequenced in this study. | ||||
The nucleotide composition of mitogenome sequences was computed with MEGA X (Hachioji, Japan) [37]. AT and GC-skew values were measured using the following formula: AT-skew = (A–T)/(A+T) and GC-skew = (G–C)/(G+C) [38]. Non-synonymous (dN) and synonymous (dS) substitution rates for protein-coding genes were estimated using PAML 4.9 (London, UK) [39].
Comparison of the gene order in whitefly mitogenomes was performed in CREx (Leipzig, Germany) [40] (http://pacosy.informatik.uni-leipzig.de/crex/form), with the common intervals parameter for distance measurement. In addition, mitogenome structures were mapped onto the sequence-based phylogeny to explore the potential phylogenetic information from mitochondrial gene rearrangements.
Each of the 37 mitochondrial genes was aligned separately. Protein-coding genes were aligned using codon-based multiple alignments under the MAFFT algorithm [35] implemented in TranslatorX [34], with the Invertebrate Mitochondrial genetic code. Poorly aligned sites from the protein alignment before back-translate to nucleotides were removed with Gblocks v. 0.91 (Barcelona, Spain) [41]. The options used for less stringent selection were: allowing smaller final blocks, allowing gap positions within the final blocks, and allowing less strict flanking positions. Ribosomal and transfer RNA genes were aligned individually in the MAFFT server [35] and with the “E-INS-i” iterative refinement method. Alignments were concatenated with FASconCAT-G_v1.04 (Bonn, Germany) [42]. The partition option “-l” was selected to simultaneously generate a gene partition output file for the concatenated supermatrix. This partition file can be used directly or with slight adjustment in the subsequent Maximum likelihood analysis.
Three types of concatenated datasets were used in the phylogenetic analysis: (1) PCG_nt, nucleotide alignment including 13 protein-coding genes; (2) PCG_aa, amino acid alignment including 13 protein-coding genes; and (3) PCGRNA, nucleotide alignment including 13 protein-coding genes, two rRNA genes and 22 tRNA genes.
Phylogenetic trees were constructed using Maximum likelihood (ML) and Bayesian
inference (BI) approaches. ML analyses were performed using IQ-TREE 1.6.10
(Vienna, Austria) [43]. Models for ML analyses (Supplementary Table 1)
were chosen for each concatenated dataset using ModelFinder (Canberra, Australia)
[44] implemented in IQ-TREE 1.6.10 [43]. For the nucleotide dataset of
protein-coding genes, data blocks were predefined by gene and codon. For the
amino acid dataset of protein-coding genes, data blocks were predefined by gene
alone. The 22 tRNA genes were defined as a single partition due to very short
gene length of each tRNA gene (60~70 bp), while two rRNA genes
were defined as separate partitions. We used the option of allowing partitions to
have different speeds (-spp). Nodal support values (BS) were evaluated using 1000
ultrafast bootstrap replicates [45]. BI analyses were conducted using PhyloBayes
MPI on XSEDE (1.8c) (Montréal, Québec, Canada) [46], as implemented in
the CIPRES Science Gateway [47]. The CAT-GTR model was employed for the
nucleotide datasets PCG_nt and PCGRNA, while the CAT-mtZOA model was used for
the amino acid dataset PCG_aa. All BI analyses involved two chains, with a total
length of 10,000 cycles. Constant sites were removed. Convergence between the two
chains was evaluated by examining the difference in frequency for all of their
bipartitions (maxdiff
A total of 197,079,804 clean read-pairs were obtained from the sequence library. GetOrganelle produced a 15,876 bp contig, while Geneious generated a 14,774 bp contig. For the mapping assembly conducted by Geneious, 28,046 of 197,079,804 reads were assembled, and the “Pairwise % Identity” reached 99.6%. The mean coverage of 14,774 bases was 286.2, with a standard deviation of 63.9. The minimum coverage was 2 and this occurred at the start site of assembly, while the maximum coverage was 448.
Alignment showed that the Geneious assembly was missing a region compared to that obtained from GetOrganelle. After annotation through MITOS, this missing region was found to span the majority of the rrnS gene and the first control region. With the exception of this missing region, Geneious presented an identical sequence to that from GetOrganelle. Therefore, the GetOrganelle assembly was used as the preferred result.
The A. psidii mitogenome contained the complete set of 37 genes usually present in insect mitogenomes. In addition, duplication of the tRNA gene cluster -trnQ_trnI_trnM was found in the control region. The mitochondrial gene order of A. psidii appeared to be unique among whiteflies. Notably, the relative position of nad1, which was nested within the control region, differed from that seen in all other whitefly mitogenomes. Moreover, the adjoining trnI and -trnQ, and the adjoining trnC and trnY exchanged positions with each other. The entire mitogenome had an AT content of 78.6%. On the major strand, the AT- and GC-skew values were –0.239 (higher T than A content) and 0.243 (higher G than C content), respectively.
A. psidii was unique in that additional start codons were used for protein-coding genes: GTG for atp6, GTT for atp8, and TTG for nad2. The remaining protein-coding genes used ATN (ATG, ATT and ATA) as the initiation codon. Eleven of the 13 protein-coding genes terminated with TAA. The abbreviated stop codon T was found for cox1 and nad1. Estimation of the evolutionary rates of protein-coding genes showed that the mitogenome of A. psidii evolved at a low rate (dN = 0.3117) relative to other whitefly species.
The program MITOS was able to locate 20 tRNA genes. trnS1 and
trnL1 were identified by alignments with other closely related species.
Secondary structures were predicted for all tRNAs (Fig. 1). A standard cloverleaf
structure was inferred for 18 tRNA genes. trnC and trnA had
T
 Fig. 1.
Fig. 1.The secondary structures of tRNA genes inferred for Aleuroclava psidii.
The rrnL and rrnS genes were located on the minor strand, with lengths of 1191 bp and 748 bp, respectively. Both these genes had very similar secondary structures to those of D. yakuba. The rrnL gene of A. psidii contained five domains (I–II, and IV–VI) with 40 helices (Fig. 2). The rrnS gene had three domains (I, II, and III) composed of 27 helices (Fig. 3).
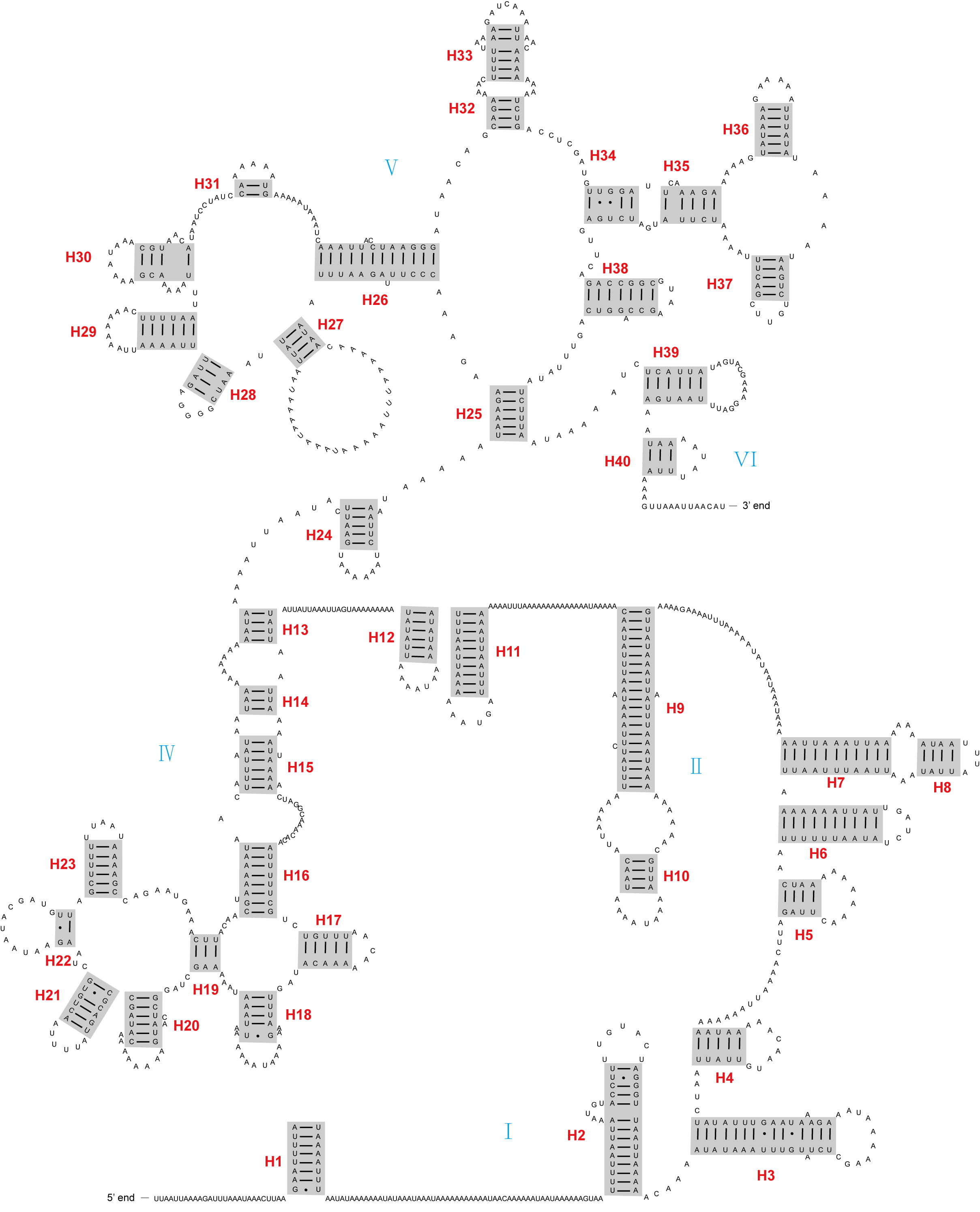 Fig. 2.
Fig. 2.The secondary structure of rrnL inferred for Aleuroclava psidii. Blue roman numbers denote the domains, while red numbers denote the helices.
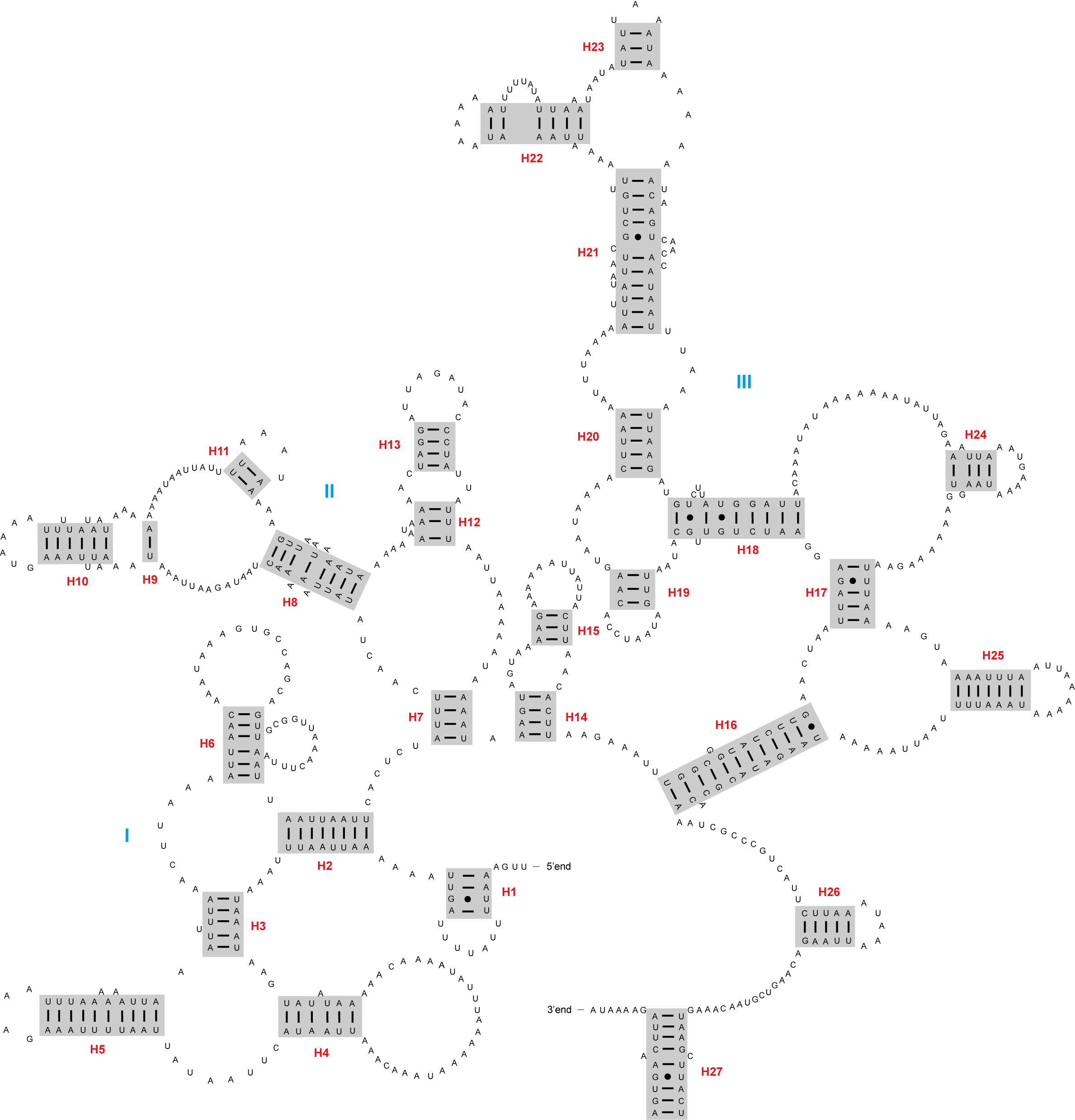 Fig. 3.
Fig. 3.The secondary structure of rrnS inferred for Aleuroclava psidii. Blue roman numbers denote the domains, while red numbers denote the helices.
The topologies resulting from ML and BI analyses were relatively concordant for the three concatenated datasets. Both Aleurodicinae and Aleyrodinae were monophyletic in all of these topologies with robust support (BS = 100, PP = 1). BI analyses of the datasets PCG_aa and PCG_nt resulted in an identical tree topology (Fig. 4, and Supplementary Figs. 1–3).
 Fig. 4.
Fig. 4.Phylogenetic tree inferred from the dataset PCG_aa using PhyloBayes under the model CAT-mtZOA (left), and comparisons of the gene order of whitefly mitogenomes (right). In the phylogenetic tree, the numbers around nodes indicate posterior probabilities. The scale bar represents substitutions/site. Bold denotes the newly sequenced species. The branch of Aleurodicus dispersus is depicted as half of its original length. Red indicates the mitochondrial gene rearrangements found in the whitefly species examined.
The main difference between the analyses was the placement of Trialeurodes vaporariorum and Neomaskellia andropogonis. On all BI trees, T. vaporariorum was recovered as the sister to all other Aleyrodinae. N. andropogonis was the sister to a clade comprising Aleurochiton and Pealius. In contrast, the phylogenetic placements of T. vaporariorum and N. andropogonis varied across ML analyses.
A sister relationship between Singhiella and Bemisia was
recovered in all BI analyses (PP = 0.99 or 1) and in ML analysis of PCG_aa (BS =
100). At the genus level, the monophyly of Aleurodicus,
Pealius, Aleurocanthus and Bemisia was supported in
most analyses. The newly sequenced A. psidii was sister to
Aleyrodes shizuokensis on the trees from PCG_aa and PCG_nt (BS
The gene order in the mitogenomes of three psyllid species is identical to that of the ancestral insect mitogenome (Fig. 4). One of these (Arytainilla spartiophila) was selected to make pairwise comparisons with the mitogenomes of whiteflies using CREx analyses. All whitefly mitogenomes investigated in this study had experienced gene rearrangements. The newly sequenced A. psidii had three rearrangements, involving transpositions of trnQ, trnY, and the protein-coding gene nad1. Notably, the nested position of nad1 within the control region was very different to that observed in other whiteflies. At least one gene rearrangement was detected in other whitefly mitogenomes. The gene order in Aleurodicus dugesii was identical to that of Arytainilla spartiophila, except for the altered position of trnY. In contrast, the gene order was highly rearranged among representatives of two clades. One clade included Aleurochiton and Pealius, and the other contained Singhiella and Bemisia. Based on CREx analyses, the distinct gene order involving altered positions of protein-coding genes in Aleyrodidae sp., Aleurocanthus spiniferus, Aleurocanthus camelliae, S. simplex, B. afer, B. tabaci and B. emiliae may have evolved from the ancestral gene order following a tandem duplication random loss event.
Despite the significant economic importance and worldwide distribution of many well-known pests belonging to the family Aleyrodidae, few studies have investigated the phylogeny of this group. Here, we obtained the complete mitogenome sequence of A. psidii, and investigated phylogenetic relationships within Aleyrodidae. These results demonstrate the power of mitogenome sequences for establishing the relationships of whiteflies. The present mitogenome data clearly resolved Aleyrodidae into two monophyletic groups, namely the subfamilies Aleurodicinae and Aleyrodinae. All genera with more than two exemplars included were monophyletic, with the exception of Aleurochiton. The close relationship of Singhiella to Bemisia was confirmed, with both forming a sister group of a clade comprising Tetraleurodes and Aleurocanthus. This arrangement concurs with an earlier study of mitogenomes [48].
The phylogenetic incongruence observed here was mainly due to the unstable positions of T. vaporariorum and N. andropogonis. The long branches leading to T. vaporariorum and N. andropogonis highlight the rapid evolutionary rate observed in both species compared to other whiteflies. Moreover, one of the Aleurodicus species, namely Aleurodicus dispersus displayed the longest branches on all ML trees. Evolutionary rate analysis indicated the substitution rates for Aleurodicus dispersus, T. vaporariorum and N. andropogonis were higher than those of all other whitefly species (Table 2). In addition to estimating the evolutionary rate from the concatenated 13 protein-coding genes, we also calculated the evolutionary rate of each of the protein-coding genes in mitogenomes (Supplementary Table 2). The results showed that Aleurodicus dispersus, T. vaporariorum, N. andropogonis and S. simplex had higher non-synonymous substitution rates. In particular, Aleurodicus dispersus had the highest dN values for the cob, nad1, nad2, nad4, nad4l, nad5 and nad6 genes. Therefore, the different positions of T. vaporariorum and N. andropogonis were likely to be the result of long-branch attraction, especially in the analyses using the homogeneous model.
| Species | dN | dS |
| Aleuroclava psidii | 0.3117 | 4.8711 |
| Pealius machili | 0.3139 | 5.0625 |
| Bemisia tabaci | 0.3154 | 5.0261 |
| Bemisia sp. | 0.3158 | 5.0574 |
| Bemisia emiliae | 0.3183 | 4.9536 |
| Aleyrodes shizuokensis | 0.3191 | 4.6099 |
| Aleurochiton aceris | 0.3193 | 5.2542 |
| Pealius mori | 0.3226 | 4.8338 |
| Aleurodicus dugesii | 0.3279 | 5.0654 |
| Aleurocanthus camelliae | 0.3352 | 4.7860 |
| Aleurocanthus spiniferus | 0.3383 | 4.8478 |
| Aleyrodidae sp. | 0.3442 | 4.4731 |
| aff Aleurochiton sp. | 0.3455 | 4.9009 |
| Bemisia afer | 0.3491 | 4.3909 |
| Tetraleurodes acaciae | 0.3538 | 4.5784 |
| Singhiella simplex | 0.3701 | 4.2424 |
| Neomaskellia andropogonis | 0.3793 | 5.1452 |
| Trialeurodes vaporariorum | 0.3829 | 3.9358 |
| Aleurodicus dispersus | 0.5338 | 5.1293 |
| Note: dN, non-synonymous substitution rates; dS, synonymous substitution rates. | ||
To further investigate the effect of long-branch attraction, we excluded the long branched Aleurodicus dispersus and reran ML analysis based on the PCGRNA dataset. This resulted in the long-branched N. andropogonis being in a different part of the tree and having a relatively close relationship to the clade including Aleurochiton and Pealius (Supplementary Fig. 4). This analysis confirms the potential attraction between Aleurodicus dispersus and N. andropogonis.
In contrast to ML analyses, the phylogenetic positions of T. vaporariorum and N. andropogonis were stable in the Bayesian inferences under the heterogeneous model. T. vaporariorum was retrieved as the first diverging clade in all BI analyses, while N. andropogonis was consistently placed as sister to a clade comprising Aleurochiton and Pealius. This result demonstrates the power of the heterogeneous model in suppressing long-branch attraction artefacts when using mitogenome sequence data to construct whitefly phylogeny.
Previous studies have suggested that mitochondrial gene rearrangements can be useful for resolving relationships within some taxonomic groups of insects [12, 49, 50, 51, 52, 53, 54, 55]. However, mitochondrial gene rearrangements are rare in Hemiptera. Nevertheless, the mitogenomes of single hemipteran groups often exhibit peculiar evolution that deserves careful examination. In this regard, whiteflies are among the most notable Hemiptera taxa, due to several interesting features. Most strikingly, several prior analyses of mitogenome sequences have reported that whitefly mitogenomes often have higher substitution rates than those of other hemipterans [56, 57]. In fact, the evolutionary rate of whitefly mitogenome sequences is too high to be used in some phylogenetic analyses [58]. In addition, whiteflies show an unpaired degree of mitochondrial gene rearrangements. Previous studies suggested that high rates of mitochondrial DNA substitution and of mitochondrial gene rearrangement were positively correlated in some insect groups [59, 60]. In the present work, we mapped the gene orders of mitogenomes onto the Aleyrodidae phylogeny based on analysis of amino acid sequences (Fig. 4). Comparisons of the gene order of mitogenomes evidenced the clade-specific evolutionary trend in whiteflies.
The mitogenome structures shown in Fig. 4 illustrate the evolutionary divergence patterns of mitochondrial gene rearrangements that frequently occur in the family Aleyrodidae. Originally, the trnW_-trnY_-trnC organization was retained in the ancestral mitogenome. However, in the new mitogenome of A. psidii, the -trnY gene was located between the trnW and -trnC genes. This pattern was shared by most whiteflies included in the present study. In addition, the tRNA genes located within the tRNA gene clusters and originally situated between the nad3 and nad5 genes and between the control region and nad2 genes can often swap positions in some taxa of Aleyrodidae. In particular, rearrangement of the tRNA gene cluster originally situated between the nad3 and nad5 genes can serve as a synapomorphy for several clades within the group. Besides the rearrangements of the tRNA genes mentioned above, the position of nad1 also changed in the mitogenome of A. psidii. This protein-coding gene was originally situated between the trnS2 and -trnL1 genes in the ancestral mitogenome. However, in the A. psidii mitogenome the nad1 gene was located between two control regions. This pattern was distinct from all other whitefly mitogenomes. Assembly from two programs (Geneious and GetOrganelle) resulted in the same position for the nad1 gene. Nevertheless, further sequencing of mitogenomes from the genus Aleuroclava is needed to confirm this result.
Aleurodicus dugesii displayed the closest gene order to the ancestral mitogenome, with only a single transposition of the -trnY gene detected in the Aleurodicus dugesii mitogenome. The trnW_-trnY_-trnC arrangement was generally seen across the entire family Aleyrodidae. Reverse transposition of the -trnL1_-rrnL_-trnV_-rrnS gene cluster to the unique gene arrangement rrnS_trnV_rrnL_trnL1 was found in Aleurodicus dispersus.
Similar to Aleurodicus dugesii, several early-diverging species within Aleyrodinae also had a small cluster of gene rearrangements, namely T. vaporariorum, Aleyrodes shizuokensis and A. psidii. The trnQ_trnI_trnM gene order was shared by T. vaporariorum, Aleyrodes shizuokensis and A. psidii, which may indicate a close relationship for these three species. In comparison, the remaining Aleyrodinae whiteflies had more extensively rearranged mitogenomes. In addition to the frequent rearrangements of tRNA genes, some of the gene arrangements involved protein-coding genes and/or rRNA genes. For N. andropogonis, the gene cluster cox3-trnG-nad3 originally located between atp6 and trnA and encoded on the major strand was translocated to a position between trnP and the control region, as well as being encoded on the minor strand. Furthermore, the rrnS gene was reversed from the typical minor strand to the major strand. Relative to D. yakuba, we identified an inversion in four species of Aleurochiton and Pealius, namely -trnR_-trnA_-nad3_-trnG_-cox3_-trnN_-trnQ located between trnS2 and -nad1. This could serve as a potential synapomorphy for the clade containing the two genera. The reverse position of trnS1 shared by N. andropogonis, Aleurochiton and Pealius may indicate a close relationship of N. andropogonis to Aleurochiton and Pealius.
The gene cluster -nad3_-trnG_-cox3 situated between -rrnS and the control region may be used as a character in support of the clade including T. acaciae, Aleyrodidae sp. and two species of Aleurocanthus. The -trnQ_-trnV cluster located between -rrns and -nad3 together with the -trnR_-trnD cluster between cox3 and the control region in T. acaciae, as well as the -trnN_-trnQ_-trnV_-trnR_-trnA cluster between -rrnS and -nad3 in Aleyrodidae sp. may be autapomorphic for these two species. The pattern of -trnN_-trnQ_-trnV_-trnR_-trnA located between rrnS and nad3 may be a synapomorphy for the genus Aleurocanthus.
The gene cluster -trnS1_-trnE_-trnF lying between atp6 and -nad5 and the gene cluster of -trnV_-trnD_-trnQ_-rrnS_-trnN_-trnR_-trnA_-nad3_-trnG_-cox3 located between -rrnL and the control region were shared by S. simplex and three species of Bemisia (B. afer, B. tabaci and B. emiliae). The transposition of -trnI and -trnW may be autapomorphic for S. simplex.
The mitogenome of A. psidii is highly rearranged and has many distinguishing characters. It has two main non-coding regions and a genome structure that is different to all other whitefly species studied to date. Our phylogenetic analyses demonstrate the potential to resolve major phylogenetic relationships within Aleyrodidae by using structural rearrangements such as those described above. Despite this, we acknowledge that taxon sampling in this work was still limited by the mitogenome sequences available for the group. Future research should aim to sequence more whitefly species from the genera Aleurodicus, Aleuroclava and Neomaskellia in order to confirm the pattern of gene rearrangements detected in this study.
mitogenome, mitochondrial genome; mtDNA, Mitochondrial DNA; PCG, protein-coding gene; rRNA, ribosomal RNA; tRNA, transfer RNA; atp6 and atp8, ATP synthase subunits 6, and 8; cob, Apocytochrome b gene; cox1-3, Cytochrome c oxidase subunits 1–3 genes; nad1-6 and nad4L, NADH dehydrogenase subunits 1–6, and 4 L genes; rrnL, large subunit ribosomal RNA; rrnS, small subunit ribosomal RNA; nt, nucleotide; aa, amino acid; dN, non-synonymous substitutions; dS, synonymous substitutions; ML, Maximum likelihood; BI, Bayesian inference; BS, bootstrap; PP, posterior probabilities.
NS, RB and HM designed the research study. NS and HZ performed the research. RB provided the specimens and performed morphological identification. NS and HZ analyzed the data. NS, HZ and HM wrote the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
This study was carried out in full compliance with the laws of the People’s Republic of China. No permissions were required for insect samples collections. The study species is not included in the ‘List of Protected Animals in China’.
We thank three anonymous referees for constructive comments that improved this paper.
This study was supported by funds from the National Natural Science Foundation of China (U1904104), Science and Technology Innovation Fund of Henan Agricultural University (KJCX2019A10).
The authors declare no conflict of interest.